
MEDICINA |
|
|
Enfermedades raras

Una historia de observación
Hace poco, a comienzos de Octubre de este año, la doctora francesa Charlotte Dravet viajaba a Madrid para recibir el Premio Dravet 2019 de la mano de la Fundación Síndrome de Dravet por su contribución al conocimiento e investigación de la enfermedad durante la celebración de la reunión anual de familias de la fundación del mismo nombre.
Obviamente la coincidencia del apellido no era casualidad y detrás está la historia de una excepcional psiquiatra y epileptóloga que le ha valido, a lo largo de su vida, infinidad de reconocimientos y mucho más que ser conocida como la descubridora de un síndrome devastador.
Dravet era a finales de los años 60 del siglo pasado una sobresaliente graduada de la Universiad Aix-Marseille, actualmente la mayor universidad del mundo francófono y por aquel entonces una casi idílica universidad en la Provenza, al sur de Francia. Con más de 5 siglos de historia, fundada en 1409 por Luis II de Anjou, se respiraba el esfuerzo de muchas naciones europeas de posguerra por investigar y progresar en el marco del final de aquella década convulsa y optimista.
La debilidad de Charlote eran los niños, así que entre 1962 y 1965 se formó en pediatría y finalmente llegó la hora del doctorado. Cualquier estudiante de pediatría conoce desde muy tempranas fechas el gravísimo problema que supone la epilepsia infantil y las trágicas consecuencias que puede acarrear. Dravet, nunca fue una mujer de elegir caminos fáciles y cómodos, así que su trabajo no iría sobre la nutrición de las papillas o las erupciones cutáneas, dado que eligió hacer su tesis sobre el síndrome de Lennox-Gastaut, una grave encefalopatía epiléptica de la infancia (el propio descubridor, que le daba nombre trabajaba en aquella universidad). 
Entrevista a Charlotte Dravet en el Décimo Congreso de Epileptología en Londres 2012 - Epilepsy Action
Una vez terminado su doctorado en 1971 fue certificada como psiquiatra y en 1972 se formó en el Departamento de EEG pediátrica del propio hospital y en el Departamento de Neurocirugía Funcional del Hospital Sainte-Anne de París. Tras conocer de primera mano en aquellos años las dramáticas consecuencias de la epilepsia en la infancia se especializó en el Centro Saint Paul de Marsella, donde trabajó hasta el año 2000 (Ya como director Médico Asociado entre 1989 a 2000) y colaboró allí con grandes especialistas como el propio Henri Gastaut , Joseph Roger o el psiquiatra pediátrico René Soulayrol. Por aquel entonces, era la doctora residente, de hecho, trabajó en aquella institución apasionadamente durante toda su carrera profesional, hasta su jubilación con la llegada del nuevo siglo, (aunque nunca ha dejado desde entonces de preocuparse por los pacientes o seguir promocionando la investigación para combatir estas desastrosas variantes de la epilepsia).
Así pues, en aquel entorno tuvo la oportunidad de acompañar y observar a los pacientes hospitalizados día y noche durante muchos años, gracias a lo cual, en 1978 y por primera vez en la literatura científica, describe la enfermedad gracias a la identificación de un grupo de pacientes con una sintomatología común.
Desde entonces ha sido mundialmente conocida por el síndrome que lleva su nombre, pero a lo largo de toda su carreara, además la Dra. Dravet ha realizado contribuciones impresionantes a la comprensión de los correlatos clínicos y electrofisiológicos de los síndromes epilépticos de la infancia. Además de su experiencia clínica y logros de investigación, la Dra. Dravet también es famosa mundialmente por su actitud humilde, su trabajo incansable y su dedicación a la epilepsia, incluyendo su esfuerzo el apoyo a numerosas asociaciones algunas formadas por familias de pacientes con síndrome de Dravet, así como abogar por una mejor atención para los niños con epilepsia. Ha producido muchas publicaciones para la comunidad, particularmente para los padres de niños con epilepsia y poder ayudarlos a enfrentar las diferentes características de la enfermedad. Pocas veces resulta imprescindible comenzar el relato sobre una patología sin realizar un apunte tan remarcado hacía su descubridor, pero sin duda este lo es. Conozcamos ahora este extraño síndrome. 
¿Cuántos afectados existen?
Se sospecha de muchos casos sin diagnosticar en España

Desde 1989 la Liga Internacional contra la Epilepsia (ILAE) lo incluye dentro del apartado de ‘Epilepsias y síndromes indeterminados respecto a la localización con crisis generalizadas y focales’.
Hoy en día todavía es una pregunta difícil de responder, el Síndrome no se describió hasta finales de 1970 y hasta 2003 no existió un test genético que ayudara a diagnosticar la enfermedad, el cual , para empeorar la situación, todavía no se aplica en todos los casos o se dificulta su acceso por diversas razones (incluidos los recortes en atención sanitaria pública o los cambios en normativa que hacen dependiente el diagnóstico por ejemplo de la calidad asistencial de las diferentes comunidades en el caso de España)
Por estas razones el número de afectados no se conoce con exactitud, aunque se estima que la incidencia de la enfermedad ronda de 1/16.000 a 1/40.000 nacimientos, lo que la encuadra de todas maneras en el grupo de enfermedades raras (ya que afecta a menos de 1 de cada 2.500 nacimientos).
En España según los últimos estudios, se calcula que hay entre 348 y 540 pacientes correctamente diagnosticados y se ha observado que un 25% de los pacientes presenta una historia familiar de epilepsia. Pero los datos de prevalencia de la enfermedad llevan a pensar que el número de pacientes diagnosticados debiera ser superior a 2.000 lo cual podría indicar un preocupante infradiagnóstico en nuestro país. 
Síndrome de Dravet en la actualidad, avances en genética molecular
Aunque normalmente deberíamos comenzar por otros apartados a la hora de hablar de un síndrome o patología, en este caso resulta imperativo hacer un inciso previo en el apartado sobre los avances en genética reciente, pues son la base que garantiza un diagnóstico a tiempo y las probabilidades de un tratamiento con éxito. Actualmente el Síndrome de Dravet está perfectamente descrito y clasificado, desde 1989 la Liga Internacional contra la Epilepsia (ILAE) lo incluye dentro del apartado de ‘Epilepsias y síndromes indeterminados respecto a la localización con crisis generalizadas y focales’. 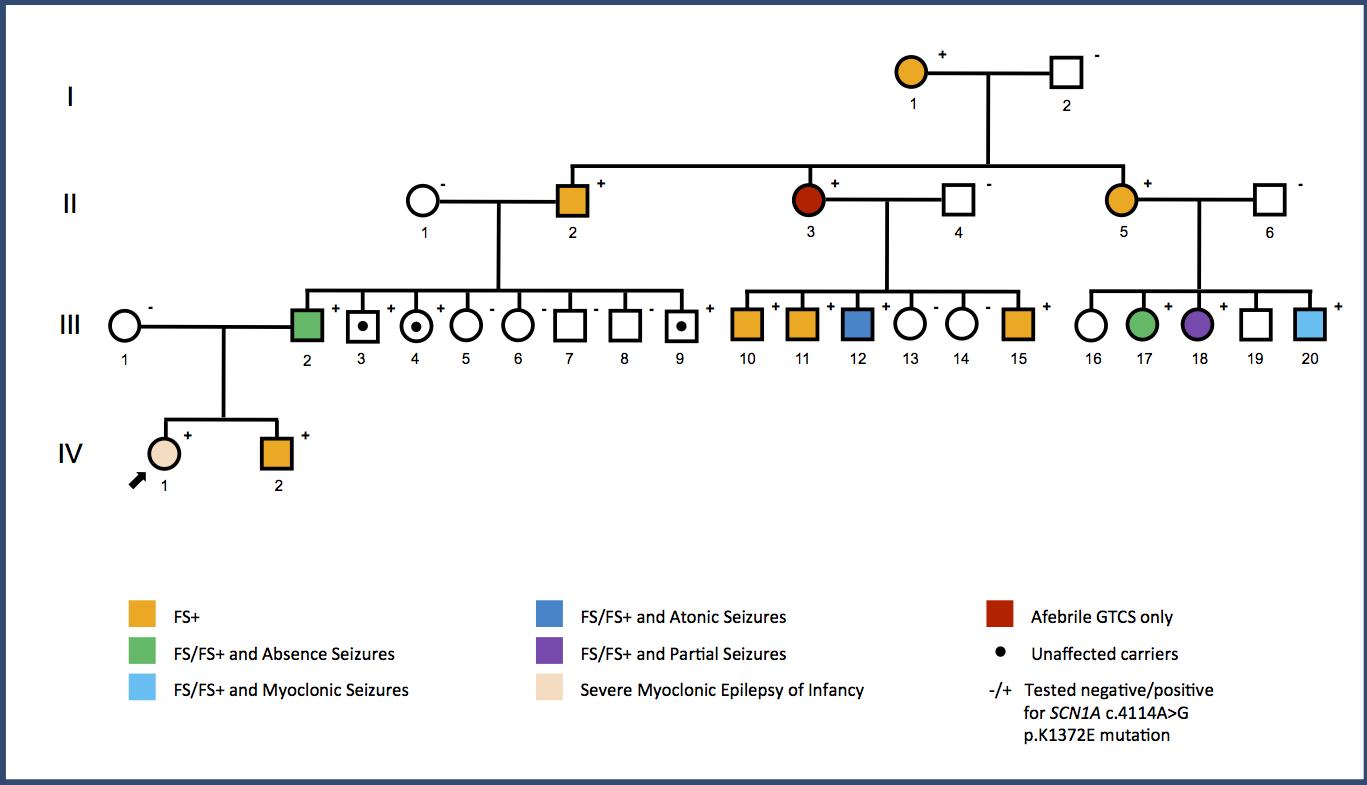
En la tabla vemos el “Pedigrí” de la gran familia GEFS +. Una mutación sin sentido da como resultado una amplia gama de fenotipos que abarcan los portadores no afectados, los fenotipos FS + y el Síndrome de Dravet. Por ejemplo, la variedad de fenotipos observados en esta familia sugiere que SCN1A no es el único causante. Habrá que investigar si los factores modificadores son genéticos o no genéticos. El patrón de fenotipos sugiere algunos factores intrafamiliares. Imagen propiedad de: The ILAE genetics comission blog – International League Against Epilepsy
En los últimos 10 años, gracias a los avances en genética molecular se ha podido identificar distintas alteraciones en el gen SCN1A como causa del síndrome de Dravet en el 80% de los casos. Así pues, con un diagnóstico precoz efectivo, cuando el niño presenta síntomas clínicos, se puede confirmar el síndrome de Dravey mediante un test genético para, a continuación, ofrecer un tratamiento muldisciplinar temprano. Se ha conseguido delimitar con precisión el diagnóstico clínico: La edad de aparición se sitúa entre los 4 y 12 meses de vida, caracterizándose por convulsiones clónicas o tónico-clónicas generalizadas o unilaterales de duración prolongada tanto en un contexto febril o en ocasiones en ausencia de fiebre.
Con edades un poco más avanzadas, es frecuente la aparición de otro tipo de crisis, como mioclonías, ausencias atípicas y parciales complejas. Pero a partir del segundo o tercer año pueden aparecer otras comorbilidades (trastornos adicionales) como el retraso del desarrollo y los EEG anormales que a menudo no son evidentes hasta esas edades. 
Diagnóstico y confusiones
Por desgracia en el primer año de vida, especialmente durante los primeros episodios, muchas veces se confunde el Síndrome de Dravet con convulsiones febriles. Hasta hace poco (primera década del nuevo siglo), el diagnóstico sólo era posible entorno a los 2-4 años de vida debido a la necesidad de esperar la evolución para poder determinarlo, pero gracias el desarrollo del test genético, el estudio molecular y el mejor conocimiento de la clínica se ha podido adelantar. Esto puede ser un problema muy grave, pues la divergencia entre una y otra es relevante; en el caso de las convulsiones febriles no se trata de epilepsia, por lo que no está indicado un tratamiento crónico con fármacos antiepilépticos. Por el contrario, en el síndrome de Dravet, es importante que el paciente sea tratado adecuadamente por un especialista con la menor demora posible. requiriendo d medidas de prevención de la fiebre, cobertura antiepiléptica y tratamiento agresivo inicial ante la posibilidad de un estado epiléptico, así como la puesta en marcha de un programa de atención temprana,. Es cierto que una primera crisis con fiebre en el síndrome de Dravet puede ser similar a una convulsión febril para un especialista poco competente o sin familiarizar con la patología, pero existen algunas particularidades que permiten sospecharlo con relativa facilidad en un profesional cualificado: Las crisis febriles del síndrome de Dravet aparecen en múltiples ocasiones antes de los 7 meses y tienden a ser prolongadas, repitiéndose en períodos breves, suelen ser hemiclónicas, (las clónicas bilaterales o generalizadas tampoco son infrecuentes) y la temperatura que las desencadena puede no ser excesivamente elevada. Aunque hemos citado el test genético para el diagnóstico certero, desafortunadamente existe aproximadamente un 20% de los pacientes donde no se encuentra la mutación genética en el gen SCN1A y puede provocar dramáticos problemas de tratamiento si no se está atento al resto de los problemas comunes que pueden facilitar un diagnóstico temprano. Los síntomas clínicos en el síndrome de Dravet se caracterizan por una epilepsia severa resistente al tratamiento que presenta las siguientes características clínicas y electroencefalográficas:
En la mayoría de los casos, las crisis epilépticas comienzan en el primer año de vida. Las primeras crisis están relacionadas con la aparición de fiebre y son convulsiones generalizadas tónicas-clónicas o unilaterales. En muchas ocasiones estas crisis desembocan en estatus epilepticus (episodios de larga duración o con repetición sin recuperación de la conciencia). Con el tiempo, también aparecen crisis afebriles o relacionadas con otros estímulos, y otro tipo de convulsiones como las mioclonías, ausencias atípicas y crisis parciales-complejas. A partir del segundo año se empiezan a observar síntomas de retraso en el desarrollo cognitivo y psicomotor. En muchos casos se observan ataxia, trastornos incluidos dentro del espectro autista, problemas alimenticios, de crecimiento y trastornos del sueño. El habla suele ser una de las facultades más afectadas.
Aún así podemos resumir una tabla con los aspectos a tener en cuenta en el diagnóstico del síndrome de Dravet:
Así, ante la mínima sospecha deberemos abordar dos vías diagnósticas. El diagnóstico clínico se realiza por tanto a través de una detallada historia de las crisis convulsivas, el tipo de convulsión, edad de aparición, etc., a lo que debemos sumar pruebas diagnósticas como electroencefalogramas y tomografías computarizadas. El diagnóstico genético nos permite identificar la causa genética molecular del síndrome de Dravet, si está presente. De esta forma, los pacientes y sus familiares se pueden beneficiar de un apropiado consejo genético familiar y de un servicio de cribado prenatal si desean tener más descendencia, igualmente confirmar el diagnóstico clínico previo. Resulta por todo ello evidente que, la inclusión del diagnóstico genético en la práctica clínica habitual, combinado con el diagnóstico clínico proporcionado por los neuropediatras, permitirá establecer una rutina diagnóstica eficaz que beneficiará a los pacientes. 
Alteraciones y problemas
Como resultado de ser una enfermedad rara existe mucho desconocimiento sobre el Síndrome de Dravet. Cuando los padres reciben la dura noticia rápidamente les asaltan las preguntas y surge la angustia: “¿Vamos a ser capaces de cuidar un niño esta enfermedad?” ¿Cómo podemos ayudarle para qué pueda tener calidad de vida? ¿Cómo actuamos frente a una crisis epiléptica? Estas son sólo algunas de las preguntas que pronto surgen y es que el Síndrome de Dravet es una de las patologías más severas dentro del espectro de la epilepsia. Los niños afectados por el Síndrome de Dravet presentan habitualmente valoraciones superiores al 75 % en la minusvalía y calificaciones de gran dependiente que requieren acompañamiento constante y ayuda las 24 horas para la realización de las actividades básicas diarias, control de los desencadenantes proconvulsivos y atención durante las crisis. Es tal la gravedad de esta enfermedad, que ha sido incluida en la lista de “menor con cáncer o enfermedad grave” equiparando sus cuidados con los enfermos de cáncer o los pacientes necesitados de cuidados continuos hospitalarios.
Pese a que la evolución del síndrome de Dravet es muy particular según cada caso, las funciones cognitivas casi siempre se ven afectadas, esta gravedad y la naturaleza del deterioro cognitivo se pueden documentar mediante pruebas estandarizadas adaptadas a la edad y a intervalos a lo largo de la vida del paciente. Dicha presencia de alteraciones cognitivas importantes precisa la implicación de los profesionales de la psicopedagogía y/o neuropsicología para implantar terapias de estimulación y enriquecimiento ambiental. A ello se suman normalmente problemas de comportamiento y psicológicos que pueden afectar su vida familiar y social. Estos factores también varían de un niño a otro, pudiendo presentar rasgos autistas, alteraciones graves de la conducta, comportamientos repetitivos, hiperactividad con o sin trastorno de la atención y problemas con la interacción social, teniendo dificultades para interactuar adecuadamente con niños de su misma edad. Muchas veces la naturaleza del síndrome les hace presentar una conducta “desafiante” que puede hacerles difíciles de manejar tanto en el hogar como en público. Algunas de estas conductas desafiantes pueden estar vinculadas a una mala comunicación basada en el deterioro del lenguaje. Estos problemas suelen obligar a una vigilancia cercana del niño, sobre todo en presencia de hiperactividad, ya que pueden no ser conscientes de situaciones peligrosas o generarlas involuntariamente. La intensidad de estos problemas puede precisar de la toma de medicamentos específicos para tratar las alteraciones conductuales. Los pacientes también pueden experimentar dificultades para dormir, tanto somnolencia excesiva o dificultad para conciliar el sueño, insomnio, apneas, terrores nocturnos o despertar prematuro. Existen muchas causas que incluyen convulsiones no reconocidas o efectos secundarios de algunos de los medicamentos antiepilépticos que pueden mejorarse mediante el ajuste de la dosis.
En el plano más “físico” los problemas también son múltiples, incluyendo retraso del crecimiento, osteopenia (densidad ósea deficiente), escoliosis (curvatura de la espina dorsal), problemas con la alimentación y el apetito (mayormente consecuencia de los tratamientos con medicamentos antiepilépticos, pero los ajustes en la dosis pueden no ser fáciles debido al riesgo de más convulsiones). También problemas con la absorción de nutrientes o dificultades con el proceso de la pubertad. Son frecuentes además las alteraciones motoras y cambios posturales. Los síntomas pueden incluir trastornos en la marcha, escoliosis, deformidades del pie o de la rodilla. La marcha tiende a deteriorarse a partir de los nueve o diez años de edad, cuando los pacientes desarrollan gradualmente un patrón especial en cuclillas cuando caminan. Algunos pacientes Dravet han necesitado llevar unos DAFOS (Ortesis dinámicas diseñadas para facilitar caminar correctamente) e iniciar la marcha a los 18 meses de edad. Obviamente las habilidades motoras finas (como la escritura y el dibujo) también se ven afectadas, al igual que la lectura,. Otros problemas serán la aparición de signos de disautonomía, es decir, alteraciones en la regulación de la temperatura, sudoración, ritmo cardíaco, la circulación sanguínea y digestión. Problemas de salud dental, frecuentes Infecciones en las vías respiratorias, del aparato digestivo o de las vías urinarias (estómago, intestinos, vejiga y riñones) y problemas cardiovasculares incluyendo alteraciones e irregularidad de frecuencia cardíaca y anormalidades en la estructura del corazón. 
Las crisis epilépticas, una ruleta rusa
Se debe tener claro que las crisis ocurren debido a la propia enfermedad del paciente, que le hace propenso a tenerlas. No hay un factor que provoque irremediablemente una crisis. Sin embargo, hay factores que facilitan el tener una crisis, los llamados: n factores desencadenantes y es importante conocerlos para intentar prevenirlos al igual que los tipos de crisis y los mecanismos básicos a nivel neurológico de estas. ¿Qué son las crisis epilépticas? Las crisis o ataques epilépticos, definidos de manera sencilla, son episodios donde se alteran las funciones cerebrales en forma de hiperexcitabilidad neuronal. Durante estos episodios críticos, la señal eléctrica empleada por las neuronas para comunicarse entre sí, se propaga de forma terriblemente “caótica” y excesiva a las neuronas vecinas o según la intensidad a regiones más alejadas. Las crisis afectan a la corteza cerebral, que es donde radican la funciones cerebrales que requieren de nuestra voluntad: Nos referimos por ejemplo a: hablar, caminar, mover los músculos de forma voluntaria, entender, pensar, memorizar, o prestar atención) También esta zona es la que recibe información de los órganos de la visión, el oído, el tacto, el olfato o el gusto. Cuando se produce una crisis epiléptica, se produce una “tormenta de comunicación” y se alteran una o varias de estas funciones, por esta razón, los síntomas durante las crisis epilépticas son muy variables, aunque también nos “informan” según la función alterada de la región dentro de la corteza cerebral que ha provocado el inicio de la crisis y las funciones de las zonas donde se propaga. Normalmente las crisis suelen tener una duración breve, menos de dos minutos, a veces sólo unos segundos. Inmediatamente después de una crisis las neuronas afectadas pierden transitoriamente su función, debido al consumo desaforado de energía durante la fase de hiperexcitabilidad. Por esta razón, tras las crisis, los enfermos suelen encontrarse agotados, somnolientos y confusos, sienten debilidad o dificultad para hablar durante minutos o incluso varias horas. Pasado un tiempo, que dependerá de la intensidad y las condiciones particulares de cada paciente, las neuronas recuperaran la energía y reanudaran su función normal. 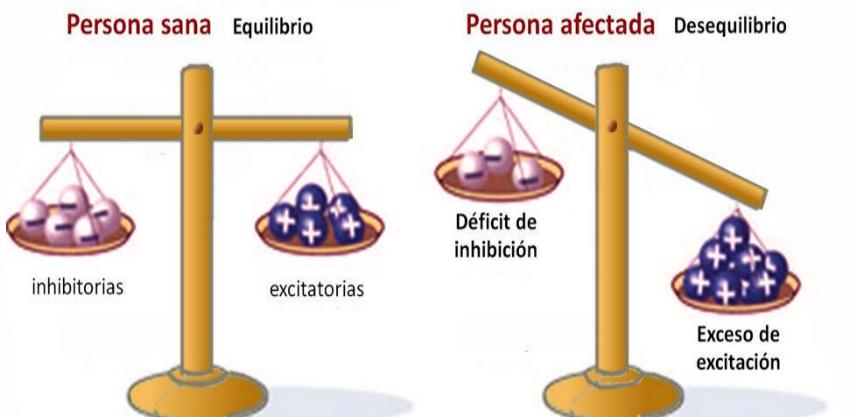
En el cerebro hay dos tipos de neuronas, neuronas inhibitorias y neuronas excitatorias, estas se comunican por impulsos eléctricos, cuando se debe detener o reducir la actividad las inhibitorias actúan como freno, pero ante un déficit de inhibición a causa de diferentes fallos en el canal de sodio, estás no podrán transmitir correctamente sus impulsos y se producirá una hiperexcitación que desencadenará una “tormenta” eléctrica, es decir, lo que llamamos una crisis epiléptica.
Estatus epilepticus El estatus epilepticus, el cual ya hemos mencionado anteriormente en este reportaje, se define como las crisis o eventos de hiperexcitabilidad neuronal que persisten por más de 30 minutos, también cuando se producen 2 o más crisis repetidas sin recuperación de la conciencia entre ellas. Por esta razón supone una emergencia neurológica que requiere una atención inmediata. La atención al paciente en estas crisis debe ser continua e intensa a lo largo de todo el evento hasta su resolución. Puede ser convulsivo o no convulsivo y la monitorización electroencefalográfica continua es de gran ayuda para el diagnóstico y para valorar la respuesta al tratamiento. Tipos de crisis Crisis convulsivas. Este tipo de crisis ocurren en todos los pacientes con síndrome de Dravet.
Crisis mioclónicas. Se producen sacudidas bruscas y breves.
Hay pacientes que no tienen este tipo de crisis. Hay pacientes que sólo las tienen al despertar, o previamente a tener otro tipo de crisis. Aparecen entre los 1 y los 5 años. Ausencias atípicas Los pacientes se quedan como embobados, con la mirada perdida. Y a veces, además de esto, presentan también mioclonías breves (parpadeo, sacudida de la cabeza). Aparecen entre los 4 meses y los 6 años, al mismo tiempo que las crisis mioclónicas. En algunos pacientes, sin embargo, aparecen más tarde, hasta los 12 años. Crisis focales Son crisis en las que sólo se afecta una parte del cuerpo.
Crisis tónicas Son infrecuentes, y consisten en rigidez brusca del tronco. Variedad de manifestaciones epilépticas En el síndrome de Dravet estas se presentan de manera diferente según cada paciente. Esto es causa de que muchas personas con síndrome de Dravet se diagnostican erróneamente con otros trastornos convulsivos como son el mencionado síndrome de Lennox-Gastaut o la epilepsia mioclónica astática, o aún peor, se les da un diagnóstico amplio de epilepsia refractaria. Síndromes de epilepsia, como el PCDH19 (una rara epilepsia ligada al cromosoma x que se encuentra con más frecuencia en mujeres que en hombres, comparten muchas características comunes con el síndrome de Dravet) Es problemático pues existen diferencias sutiles entre estos síndromes de epilepsia y Dravet que exige un detallado análisis diagnóstico, de ahí por ejemplo la utilidad del diagnóstico genético. Crisis y mortalidad En los últimos estudios la mortalidad asociada al síndrome de Dravet ha sido descrita con porcentajes alrededor del 15%, debido principalmente a la SUDEP (muerte súbita inesperada en la epilepsia), convulsiones prolongadas, accidentes relacionados con convulsiones como ahogamiento e infecciones, representando actualmente la mitad de los casos de mortalidad en el síndrome de Dravet, aunque en los últimos años, se ha producido una leve mejoría de los datos, que parece estar relacionada con un mejor manejo de las crisis y una mejor elección de los fármacos antiepilépticos. De ambos aspectos se deduce la importancia de un diagnostico correcto y temprano como esencial para el abordaje correcto de las crisis. Factores susceptibles de provocar una crisis epiléptica Algunos de los pacientes con síndrome de Dravet son sensibles a diversos estímulos ambientales que pueden desencadenar convulsiones, por lo que se debe estar atentos y prevenir situaciones desencadenantes: Patrones Visuales Los patrones visuales (repetición de formas visuales) pueden desencadenar convulsiones en algunos pacientes. Aunque no siempre es fácil evitarlos, existen algunas soluciones, como enmascarar completamente un ojo. Fotosensibilidad Algunos pacientes con síndrome de Dravet pueden tener convulsiones provocadas por luces brillantes como luces estroboscópicas o el sol reflejado desde el agua en la playa. Para otros, los colores y las formas parpadeantes pueden ser importantes, como los videojuegos o los dibujos animados. Para que no se desencadenen las crisis es aconsejable:
Temperatura El aumento de la temperatura corporal es un factor desencadenante de las convulsiones, así que conviene evitar los cambios bruscos de temperatura corporal. Evitar los deportes intensivos que conlleven fuerte elevación de la temperatura, el ambiente caluroso (estancias cerradas, con muchas personas, en el automóvil, etc), los baños muy calientes o piscinas en similares condiciones o a pleno sol. Un truco empleado por algunos pacientes consiste en utilizar chalecos de enfriamiento o neoprenos para impedir los cambios bruscos en la temperatura corporal. 
Tratamiento y protocolo en situaciones de emergencia
Cuando hablamos de crisis es importante detener las convulsiones de larga duración que puedan convertirse en estatus epilépticos, para ello comúnmente se usan las benzodiacepinas de emergencia. En los hogares de los pacientes normalmente los tratamientos de emergencia consisten en su mayoría en fármacos antiepilépticos de acción rápida. Los especialistas deben estudiar cada caso con sus particularidades para recetar el medicamento apropiado para detener las convulsiones de larga duración. Además es importante que un experto sanitario indique con precisión cómo usar los medicamentos, ya que hay que emplearlos con sumo cuidado. Existen diferentes de administración de estos medicamentos: nasales, orales o rectales Cuando un niño tiene convulsiones prolongadas que requieren tratamiento hospitalario supone un impacto y estrés importante en la familia y cuidadores. Si aparecen convulsiones graves de larga duración debe haber un plan minuciosamente elaborado con un protocolo preciso y claro, no solo para el niño con síndrome de Dravet, sino también incluso para padres y hermanos. Resulta difícil predecir cuándo puede acaecer una crisis y los padres/cuidadores deben estar siempre preparados. Cuando se sale del hogar con un paciente es muy recomendable llevar al menos los siguientes elementos:
Si los padres tienen otros hijos, deben asegurarse de tener un plan de contingencia para ellos. Hay que considerar tener un “padre de guardia” que acompañe al niño Dravet a urgencias. El otro padre o adulto designado puede quedarse con los otros niños para que puedan continuar con sus actividades. Los padres deben de ser también conscientes de cómo afectará psicológicamente la enfermedad, los cuidados, atención y situaciones de emergencia al resto de los hermanos, pues estas vivencias podrían condicionar su desarrollo de forma negativa de no tenerse en cuenta. Plantilla para situaciones de urgencia En la Fundación Síndrome de Dravet se ha desarrollado un protocolo médico de urgencia específico para el síndrome de Dravet, para hacer uso de él en el caso de que tenga una crisis prolongada. El ejemplo de protocolo médico de urgencia que ofrecen se puede enviar al neurólogo para adaptarlo y llevarlo siempre junto con el paciente con síndrome de Dravet para entregárselo al personal de urgencias en el caso de que haya una crisis prolongada. 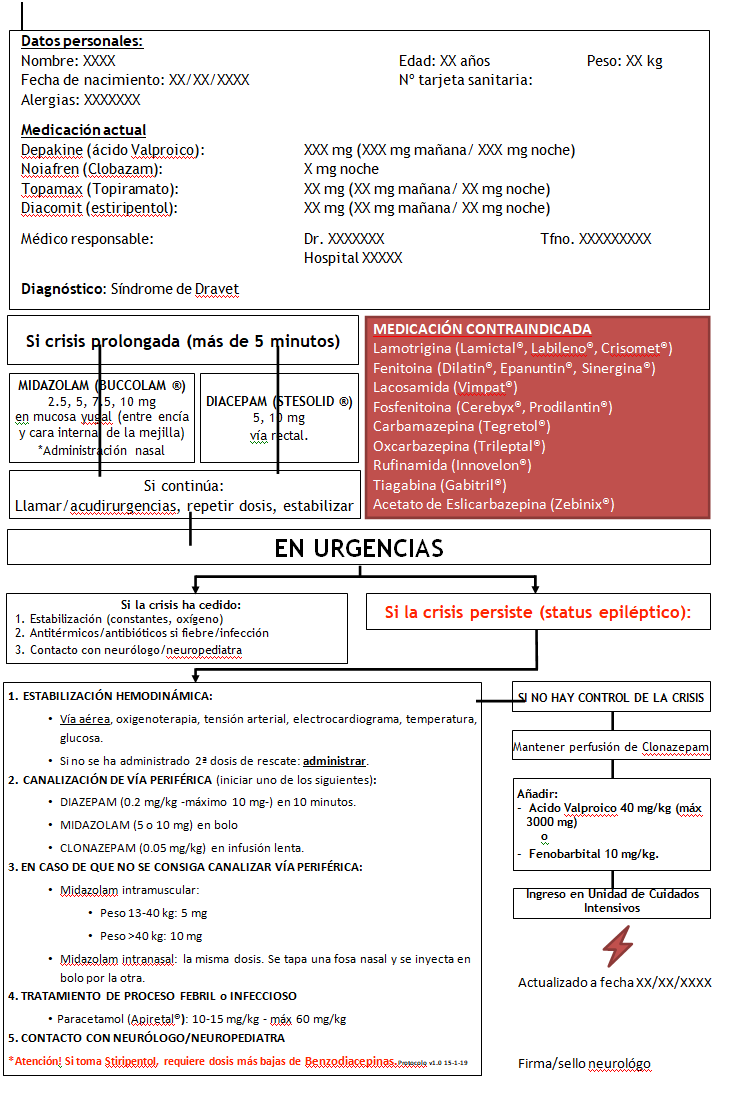
Puedes descargar el protocolo de urgencia desde la Fundación Síndrome de Dravet para adaptarla a un caso concreto pulsando aquí.
¿Cómo manejar una crisis epiléptica? Las crisis epilépticas pueden ser muy variadas como hemos dicho y con diferentes factores que las desencadenan. Así que lo esencial es estar asesorado por buenos especialistas que instruyan a los cuidadores con las medidas a tomar y no decir con elecciones arbitrarias o consejos encontrados en Internet. Una vez dicho esto, las medidas generales, veamos básicamente las medidas a tomar: Si el paciente tiene sacudidas de todo el cuerpo, lo que hay que hacer es evitar que se golpee la cabeza. Podemos sujetarla entre las manos o poner un cojín debajo. Si ha perdido la consciencia y vomita, conviene ponerlo de lado, para que no se atragante. Y si no vomita, se debe estirar suavemente el cuello y colocar la cabeza hacia atrás, para que el aire pase con normalidad. Si tiene comida en la boca, intentar sacarla para que no se atragante. Dependiendo de la medicación, se puede administrar por vía rectal, o en la mucosa oral. Tardan unos minutos en hacer efecto, pero por lo general, si han pasado 5 minutos y no ha cedido la crisis, se debe repetir la misma dosis de nuevo. En esos casos, lo más conveniente sería llamar a urgencias, porque es importante que la crisis no se prolongue demasiado. Hay algunas crisis a las que no vamos a poner tratamiento, como en las sacudidas breves (mioclonías) o en las ausencias. En otros casos podemos esperar uno o dos minutos a ver si paran solas. Pero si el paciente tiene varias crisis seguidas, o duran más de un minuto, o tienen pérdida de consciencia, se debe poner entonces el tratamiento. Por lo general, tras una crisis epiléptica, y más si se ha puesto tratamiento, el paciente se encontrará adormilado y sin fuerzas. Debemos quedarnos con él hasta que se haya recuperado. No debemos dejarle beber si sigue adormilado, porque se puede atragantar. Y no se le puede dejar solo, porque puede vomitar, o puede tener una nueva crisis epiléptica QUÉ NO HACER
Tratamiento Todavía se dispone de opciones muy limitadas a la hora del tratamiento y se requiere un cuidado constante para una persona que padece este síndrome. Este cuidado continuo afecta muy gravemente a la calidad de vida tanto del paciente como la de su familia. Para empezar, los pacientes con esta dolencia enfrentan una tasa de mortalidad alrededor del 15% debido a la SUDEP (muerte súbita inesperada en la epilepsia), las convulsiones prolongadas, accidentes relacionados con convulsiones como ahogamiento e infecciones. Así que ante un síndrome para el que no se conoce cura ni fármacos específicos y que no solo es refractaria a tratamiento con los antiepilépticos disponibles actualmente, sino que algunos de estos pueden producir síntomas más severos y de difícil control, lo primero para un tratamiento eficaz que hay que recalcar es un diagnóstico precoz que combine diagnóstico clínico y genético de ser el caso (80% de los casos). Debe quedar muy claro que todos los tratamientos deben realizarse bajo la supervisión de un médico con conocimientos específicos sobre el síndrome de Dravet. Actualmente hay muchos tratamientos antiepilépticos en todo el mundo, algunos probablemente no estén o no estén aprobados oficialmente en el país desde donde este leyendo este artículo. Especialmente con este síndrome todo cambio o modificación del tratamiento antiepiléptico debe realizarse solo con la aprobación del médico que sigue regularmente al paciente y cualquier duda o pregunta sobre los tratamientos debe realizarse a su médico especialista antes de decidir cualquier cambio. Tratamientos farmacológicos Los medicamentos antiepilépticos son un gran grupo de compuestos empleados en el tratamiento de las crisis epilépticas, la diversidad y variedad de nombres con la que se comercializan en todo el mundo es enorme. Se dividen en varias clases y su uso difiere mucho dependiendo del trastorno epiléptico específico. Algunos pueden mejorar la situación del paciente, mientras que otros de no administrarse para la condición adecuada pueden empeorarlo o incluso conducirle a un desenlace fatal. En nuestro país la disponibilidad de medicamentos específicos es bastante buena aunque depende de las regulaciones institucionales y suelen ocurrir cambios habitualmente, así como en las decisiones de comercialización del fabricante farmacéutico, pero en general la accesibilidad no ofrece grandes complicaciones. El problema está en que el síndrome de Dravet es una epilepsia altamente farmacorresistente, lo que significa que las convulsiones son difíciles de controlar y muchos pacientes se tratan con más de un medicamento antiepiléptico. Medicamentos frecuentes El valproato y las benzodiacepinas han demostrado con la experiencia ser eficaces en la reducción de las crisis de estos pacientes. Se recomienda iniciar el tratamiento con valproato tras la primera crisis. Si no se consigue control de las crisis, se recomienda agregar topiramato o bien estiripentol. Tanto estiripentol como topiramato se suelen administrar añadiendo de base valproato y clobazam. De todas maneras cuando se acude a una sale de emergencias, el médico puede usar otros tratamientos según el estado clínico del paciente, la práctica local y la disponibilidad de medicamentos, lo cual obliga a una elección con sumo cuidado. Medicamentos a evitar La lista a continuación indica algunos de los medicamentos que pueden empeorar las convulsiones y, por lo tanto, deben evitarse. Resulta muy importante que a la hora de tratar una crisis el especialista sea consciente de la idiosincrasia específica de este síndrome y las particularidades del paciente. Tratar una crisis epiléptica de Síndrome de Dravet sin saberlo puede llevar a consecuencias desastrosas.
Otras medidas antiepilépticas Los medicamentos antiepilépticos no la única manera para controlar las convulsiones y se pueden considerar otros tratamientos además de los medicamentos que han demostrado eficacia aunque con resultados variables (además no solo en el síndrome de Dravet). Hay que tener, eso sí, muy presente que cada paciente con este síndrome de Dravet es único y lo que podría funcionar para un niño puede no funcionar para otro.
La dieta cetogénica es una dieta rica en grasas, adecuada en proteínas y baja en azúcar. La dieta obliga al cuerpo a quemar grasas en lugar de azúcares. Normalmente, los azúcares contenidos en los alimentos se convierten en glucosa, que luego se transporta alrededor del cuerpo y es particularmente importante para alimentar la función cerebral. Sin embargo, si hay muy poca azúcar en la dieta, el hígado convierte la grasa en ácidos grasos y cuerpos cetónicos. Los cuerpos cetónicos pasan al cerebro y reemplazan la glucosa como fuente de energía. Un nivel elevado de cuerpos cetónicos en la sangre, un estado conocido como “cetosis”, puede llevar a una reducción en la frecuencia de las crisis epilépticas. Existen variantes de esta dieta, que incluyen recetas caseras o fórmulas preparadas para consumir. Hay que ir con mucho cuidado respecto a los consejos que se pueden encontrar en Internet y nunca iniciar ningún tipo de estas dietas sin supervisión médica.!!!
El nervio vago es un nervio especial que transporta los impulsos motores del cerebro a varios órganos (pulmones, corazón, intestinos, vasos sanguíneos…) y viceversa. La estimulación vagal puede regular los ataques epilépticos por un mecanismo de acción desconocido. La estimulación del nervio vago consiste en la implantación quirúrgica de un dispositivo debajo de la piel del tórax, similar a un marcapasos. Este dispositivo luego se conecta mediante un cable oculto debajo de la piel al nervio vago en el cuello. 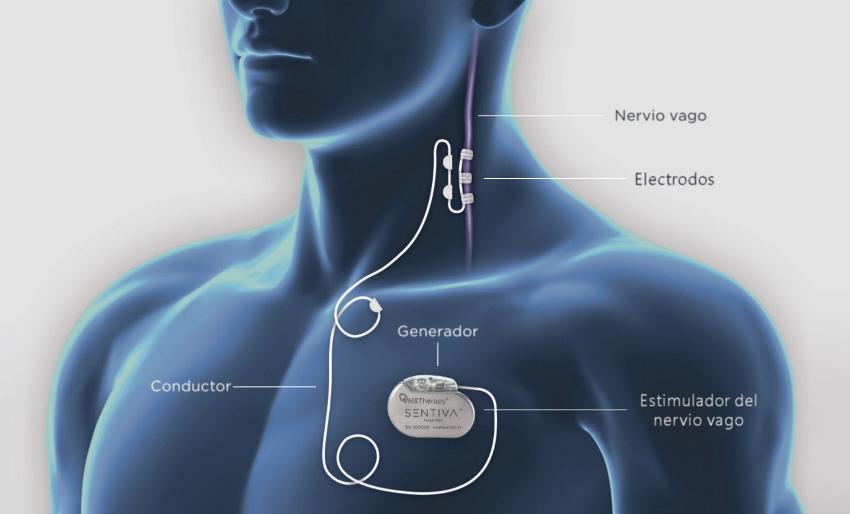
Estimulación nervio vago –Fundación síndrome de Dravet
Cuidados integrales La atención integral tiene como objetivo prevenir y/o tratar otros problemas que no sean las convulsiones y que vienen con el síndrome de Dravet: comorbilidades (presencia de uno o más trastornos además de la enfermedad primaria). El tratamiento puede no eliminar estos problemas, pero puede reducir su impacto en los pacientes y sus familias. Aunque cada afectado presenta su propio cuadro clínico algunas condiciones secundarias son compartidas por casi todos los pacientes. La existencia de estas comorbilidades exige la actuación de otros profesionales en trabajo en equipo en el abordaje integral y multidisciplinar de la enfermedad.
La fiebre es uno de los factores desencadenantes de ataques epilépticos más frecuentes. Por lo general, es bastante fácil controlar la fiebre con medicamentos. La fiebre tiene varias causas, pero la mayoría de las veces es un signo de infección. La prevención de algunas infecciones con la vacunación puede ser útil.
Existe la sensación general de que los pacientes con síndrome de Dravet son más sensibles a las infecciones en comparación con otros. Sin embargo, no tienen ninguna deficiencia inmunológica conocida. Se sabe que cada vez que una persona con síndrome de Dravet tiene fiebre, es más probable que experimente convulsiones. Si bien es cierto que algunas vacunas pueden causar una fiebre corta que puede desencadenar una convulsión, todavía se recomienda encarecidamente que los niños con síndrome de Dravet reciban todas las inmunizaciones infantiles de rutina. Una dosis correcta de medicamentos antipiréticos (por ejemplo, paracetamol o ibuprofeno) por lo general es efectiva para reducir la fiebre. Recuerde pedirle a su médico de cabecera una receta de medicamentos antipiréticos. Incluso si estos pacientes son sensibles a la fiebre, es imposible eliminar todas las enfermedades infecciosas. Deben tener una vida social lo más normal posible y no deben estar “aislados”. Recientemente, una familia ha optado por ponerle un termómetro digital al paciente Dravet y monitorizar la temperatura corporal durante 24 horas. Esta familia ha sido capaz de anticipar las crisis epilépticas al inicio de una infección y han sacado unas conclusiones muy útiles a la hora de anticipar una convulsión generada por un cambio de temperatura corporal. Para más información sobre este estudio, pinche aquí.
Los padres a menudo temen que la vacunación pueda desencadenar convulsiones, especialmente porque: -Uno de los efectos secundarios de las vacunas es a veces causar fiebre -La fiebre en algunos niños con epilepsia puede desencadenar una convulsión -El primer episodio epiléptico para el síndrome de Dravet a menudo es desencadenado por una fiebre Sin embargo, se recomienda encarecidamente vacunar a estos niños para protegerlos de enfermedades infecciosas graves.

Evolución de la enfermedad
La evolución de la enfermedad es diferente en cada paciente, pero siempre de por vida. Como ya hemos dicho los primeros signos de este síndrome son convulsiones durante el primer año de vida en un niño con un desarrollo normal. Las convulsiones varían, pero generalmente persisten durante toda la vida del paciente. Además, este síndrome afecta el desarrollo psico-motor y las funciones cognitivas. Las funciones motriz, cognitiva y de comunicación se estabilizan pero los retrasos significativos permanecen en diversos grados. Curso clínico Suele tener una evolución de crisis epilépticas dependiente de la edad, identificándose tres etapas:
El primer evento epiléptico generalmente ocurre “de la nada” normalmente entre los 4 y 8 meses de edad en un niño que se desarrolla con normalidad y no tiene antecedentes que sugieran un problema. En la mayoría de los casos, la primera crisis es inducida por una fiebre leve (37-38 ° C) a causa de una enfermedad o después de una vacunación. Por lo general, se trata de una convulsión clónica, ya sea inicialmente generalizada o solo en un lado del cuerpo (hemiclónica). Si es de un lado, puede permanecer en ese lado o extenderse a ambos lados y generalizarse. La primera convulsión suele ser una convulsión prolongada (> 15 minutos) que a veces se convierte en un estado epiléptico (> 30 minutos). Durante las próximas semanas o meses, el niño tendrá otras convulsiones, febriles o afebriles, a pesar del uso de medicamentos anticonvulsivos. En este punto, el electroencefalograma (EEG) y otras investigaciones de neuroimagen (resonancia magnética) casi siempre son normales.
Durante este período, la frecuencia de las crisis epilépticas suele aumentar. Las convulsiones pueden ocurrir con o sin fiebre, y con frecuencia incluyen episodios de estado epiléptico (> 30 minutos). Aparecen otros tipos de convulsiones: convulsiones mioclónicas, ausencias atípicas y convulsiones focales. Algunos factores ambientales pueden desencadenarlos: destellos de luz excesivos o intermitentes o patrones y diseños especiales, como patrones geométricos regulares, líneas o puntos, esfuerzo físico o incluso excitación. Durante esta fase, las convulsiones suelen ser extremadamente frecuentes, intensas y prolongadas, lo que da como resultado múltiples hospitalizaciones.
Desde la mitad de la infancia hasta la adolescencia, las convulsiones generalmente mejoran, con una reducción y, a veces, la desaparición de las crisis focales, las ausencias atípicas y las crisis mioclónicas. Las convulsiones febriles convulsivas generalmente persisten, aunque el número de fiebres es menor. El número y la duración de las convulsiones disminuyen, pero las convulsiones rara vez se detienen por completo. Estas convulsiones a menudo se agrupan, especialmente al principio o al final de la noche. En la mayoría de los pacientes, el estado epiléptico es considerablemente menos frecuente. Desarrollo psicológico y cognitivo El síndrome de Dravet tiene una evolución del desarrollo característica. El retraso en el desarrollo y la discapacidad intelectual casi siempre están presentes. Al igual que en el curso clínico de las convulsiones, se pueden identificar tres etapas, aunque las edades pueden variar:
El desarrollo psicomotor y cognitivo del niño suele ser normal durante el primer año de vida.
Entre el primer y segundo año de edad, a menudo se observa una desaceleración general del desarrollo. Aparecen trastornos intelectuales y de comportamiento. Su severidad puede variar de un niño a otro. El habla y el lenguaje son los primeros en verse afectados, pero la mayoría de las otras áreas de desarrollo se ven afectadas gradualmente. El niño está a menudo de mal humor, hiperactivo, contrario, terco y obstinado. Los problemas de comunicación dificultan aún más la interacción social. Los trastornos del sueño son comunes. Muchos de los niños tienen problemas con la coordinación, como la marcha descoordinada (ataxia) y problemas con las habilidades motoras finas (coordinación de las manos y los dedos).
Al llegar a esta fase, los trastornos psicomotores y cognitivos tienden a estabilizarse. El desarrollo puede continuar lentamente o reiniciarse si ha habido un período de regresión. La discapacidad intelectual permanente varía de moderada a severa, dependiendo de la evolución durante los años anteriores. La comunicación a menudo sigue siendo difícil y se pueden observar rasgos autistas. En general, las habilidades del habla coinciden con el nivel intelectual general del niño, pero la comprensión del lenguaje sigue siendo mejor que la capacidad para producir el habla. Ocasionalmente pueden tener un comportamiento agresivo o incluso psicosis. Desarrollo a largo plazo Como ya hemos comentado fue inicialmente descrito en 1978, hasta 1991 no se reconoció como Síndrome de Dravet y hasta 2001 no se confirmó el origen genético. Debido a ello, la evolución a largo plazo y la esperanza de vida no son bien conocidas, pero según la experiencia clínica, queda claro que el manejo temprano y apropiado del síndrome de Dravet parece dar lugar a un mejor resultado. En enero de 2019 se publico los resultados de un estudio para la determinación de la epidemiología, el flujo de pacientes y el tratamiento del síndrome de Dravet en España (Gil-Nagel, Sánchez-Carpintero, San Antonio, Mistry, Barker, Shepherd, Gil) Se efectuó un estudio Delphi de dos rondas en el que participaron 19 médicos. Los cuestionarios se basaron en una revisión de la bibliografía y fueron validados por expertos clínicos. Se alcanzó consenso si los temas se referían a la práctica clínica habitual y la experiencia individual, o si el coeficiente de variación entre las respuestas era <= 0,3. El número estimado de pacientes nuevos con síndrome de Dravet es de 73 al año. La prevalencia se calcula entre 348 y 540 pacientes y se diagnostica principalmente en niños. La supervivencia varía entre los 5 y los 60 años. No existe ningún seguimiento normalizado para los pacientes de más de 18 años de edad, y las tasas de mortalidad son inciertas. No existen guías normalizadas para diagnosticar o tratar el síndrome de Dravet. Se tarda de 9 a 15 meses en confirmar el diagnóstico, y la disponibilidad de los análisis genéticos es irregular. Normalmente se utilizan el ácido valproico, el clobazam, el estiripentol y el topiramato. La escasa eficacia y la seguridad son los motivos principales de los cambios de tratamiento. En las conclusiones se remarco que todavía la epidemiología del SD en España es poco conocida, y sigue habiendo necesidades no cubiertas en algunas áreas. Las opiniones de expertos suponen un punto de partida para poder investigar la realidad del SD en España. Los estudios epidemiológicos, los criterios de consenso, el acceso fácil a las pruebas genéticas, las opciones de tratamiento, la formación y la investigación de la calidad de vida relacionada con la salud constituyen todos ellos aspectos muy necesarios. La dura tarea del cuidador El cuidado de un paciente con el Síndrome de Dravet en el núcleo familiar supone una gran carga emocional y un fuerte desgaste: Las rutinas, los horarios, los hábitos, etc se ven gravemente condicionados y alterados. Es frecuente que los padres tengan que abandonar sus propios deseos para poder satisfacer las necesidades básicas de su hijo. El cuidado de un paciente con estas características implica brindarle un entorno seguro, educarle acorde con su nivel intelectual, adaptar las actividades de ocio a sus limitaciones… y, sobre todo, mantenerse en estado de alerta ante una posible crisis epiléptica. Estas condiciones de vida para los cuidadores llevan a que sufran un gran agotamiento físico y mental ya que la mayor parte de su día estará dedicado al cuidado del niño, acarrean costes o consecuencias negativas en la vida familiar. Al llegar al núcleo familiar un niño con Síndrome de Dravet se ven obligados a reorganizar las rutinas diarias y disponen de menos tiempo para poder estar con los demás hijos. El trabajo y la situación económica se resienten, aparecen dificultades para compaginar el trabajo y el cuidado del hijo, sufragar los gastos médicos. La manera de disfrutar del ocio cambia drásticamente y las actividades en familia se deben ajustar a las posibilidades del niño, corriendo el riesgo en muchas familias de dejar de relacionarse con otras personas a causa del trabajo y el celo protector Como en las familias de otros pacientes con enfermedades muy graves, los cuidadores sufren más dolencias musculares y cansancio físico que las personas que no tienen a su cargo una persona dependiente Afecta al estado de ánimo del cuidador y en el cuidado correcto del niño. El estado de ánimo muchas veces cambia, sobre todo, sin la asistencia sanitaria y ayudas institucionales en familias con recursos limitados, entonces, la tristeza, la irritabilidad y la desesperación son sentimientos que pueden empeorar el bienestar psicológico del cuidador. Pronto pueden aparecer las peleas de pareja y las separaciones. Para poder afrontar las dificultades del día a día muchas veces no es suficiente con la ayuda del núcleo familiar. Deben sentir el apoyo de las amistades y las instituciones públicas ya que sentirse parte de un grupo ayuda a compartir con los demás los problemas del día a día, pedir y dar ayuda, mejora la autoestima y la confianza, saber que no están solos y nos ayuda a entender que no somos las únicas personas con problemas. Además ocuparse terceros ocasionalmente del niño da un “respiro” inestimable a los padres para que puedan tener calidad de vida ellos también y “recargar las pilas” pudiendo abarcar otros aspectos de la vida esenciales para el equilibrio físico y emocional como puede ser el deporte, otro tipo de actividades personales de asueto a o simplemente poder descansar. Muchas personas no saben cómo actuar ante una persona con discapacidad intelectual, existe muy poca información real, así que observan su comportamiento sin entender por qué es de tal o cual manera y les resulta muy incómodo interactuar con ellos. Los niños con Síndrome de Dravet actúan y se comunican con los demás de una forma diferente. Se podría decir que “piensan en imágenes” y esto les impide poder hacer razonamientos muy complejos, entender ideas abstractas o anticipar hechos que los demás consideramos previsibles. Su mente se basa en acciones cortas y sencillas. Su memoria, la atención, la empatía, el habla y la socialización se ven afectadas y muchas veces la falta de comprensión de las personas hacia los síntomas hace que su comportamiento se entienda como hostil, lo cual empeora su conducta ante la incomprensión del entorno y de sus pocos recursos para poder transmitir lo que sienten. Entender por qué actúan de una manera u otra según la situación, nos ayudará a ponernos en su lugar y nos facilitará acercarnos e interactuar con ellos de una forma más efectiva. Para su familia es esencial que sientan que pueden integrarse, que son comprendidos y que no están solos, porque un niño con Síndrome de Dravet puede ofrecer cosas y sentimientos tan maravillosos como cualquier otro niño, solo debemos comprenderlos un poco y saber lo duro que es para unos padres lidiar con todo esto. Fin de la 1era parte, pulsa aquí para acceder a la 2da parte: Genética e investigación en el Sindrome de Dravet Con cariño a Maya
Para más información la Fundación Síndrome de Dravet dispone de un amplio contenido sobre la enfermedad, atención a los pacientes o sus cuidadores y recientes investigaciones.
Autor: JF Alonso
Temas relacionados: Divulgación científica, Biomedicina, Medicina, Inflexion Point Doctor, Enfermedades Raras
Reconocimientos y más información sobre la obra gráfica ADVERTENCIA: En este foro, no se admitirán por ninguna razón el lenguaje soez y las descalificaciones de ningún tipo. Se valorará ante todo la buena educación y el rigor sobre el tema a tratar, así que nos enorgullece reconocer que rechazaremos cualquier comentario fuera de lugar.
0 Comentarios
Deja una respuesta. |


Ciencia y Tecnología



Investigación Médica y Salud


Documentación a Fondo
Educación y Formación







Sociedad, Igualdad y Sostenibilidad





Cultura y Ocio










|
