

 A pocos días de celebrarse el día internacional de las enfermedades raras, desafortunadamente muchas personas tienden todavía a confundir las enfermedades raras (ER) y a la totalidad de las personas que las padecen con pacientes de psiquiatría, hipocondrías muy extremas o síntomas y síndromes muy peculiares. Para empeorar las cosas, los medios de comunicación acostumbran a poner de relieve los casos más espectaculares y amarillistas empeorando esta percepción. Vamos a recorrer con todo rigor unas y otras, al igual que la delgada línea roja que las separa, aunque debemos tener en cuenta que ambas merecen todo el respeto y la atención médica necesaria para curar al paciente. El problema radica en la facilidad con que los medios tildan a problemas del campo de la psiquiatría o enfermedades con una prevalencia relativamente baja pero con síntomas externos muy espectaculares y sensacionalistas como enfermedades realmente raras, con gran dificultad para la diagnosis o de difícil tratamiento con los procedimientos y fármacos actuales. Es cierto que a veces se produce una coincidencia entre la baja prevalencia, la espectacularidad de los síntomas y la dificultad para su tratamiento, pero no es ni de lejos lo común a la hora de citarlas normalmente, pues ante todo hoy en día, fuera del sector médico y científico desgraciadamente prima el sensacionalismo y la imagen dramática o desagradable que pueda atraer la atención rápidamente. Además, centrarse en las segundas resulta realmente sencillo, gracias al abanico increíble de trastornos que esa compleja máquina que es nuestro cerebro puede llegar a producir, veamos primero las más conocidas y que constantemente son citadas cuando se intenta hablar de “enfermedades raras” sin demasiado conocimiento de la materia, pero si mucho interés en atraer al público: Dispraxia Diagonística, Síndrome del Dr. Strangelove o de la mano extraña (CIE-9 781.8) Comenzaremos con una condición mental muy conocida y que frecuentemente se menciona equivocadamente como ejemplo de una de las enfermedades “más raras” en la cual, una de las manos de quien la padece pareciera adquirir una personalidad propia y ajena, con la capacidad de obrar por su cuenta. Obviamente es conocida por “Dr.Strangelove” en honor a la célebre película de Stanley Kubrick, protagonizada por Peter Sellers, ¿Teléfono rojo?, volamos hacia Moscú (1964), donde la mano de Strangelove intenta repetidamente realizar el saludo nazi o estrangular a su propietario que, por cierto, por dos veces se equivoca y se dirige al presidente de los Estados Unidos como “Mein Führer”… Abandonemos lo anecdótico; se describe por primera vez en 1908 por el médico alemán Kurt Goldstein, es decir ya hace más de un siglo y el síndrome es común en pacientes sometidos a una comisurotomía, una operación quirúrgica consistente en la sección del cuerpo calloso de un paciente, impidiendo la comunicación entre los dos hemisferios cerebrales. También puede ocurrir después de neurocirugías, en personas con accidente cerebro-vascular, infecciones, neoplasias, aneurismas o en pacientes que sufren enfermedades neurodegenerativas. Esto ha causado que con el paso de los años exista un elevadísimo número de casos documentados, que la hace sorprendente pero no ajena para la medicina y mucho menos rara. 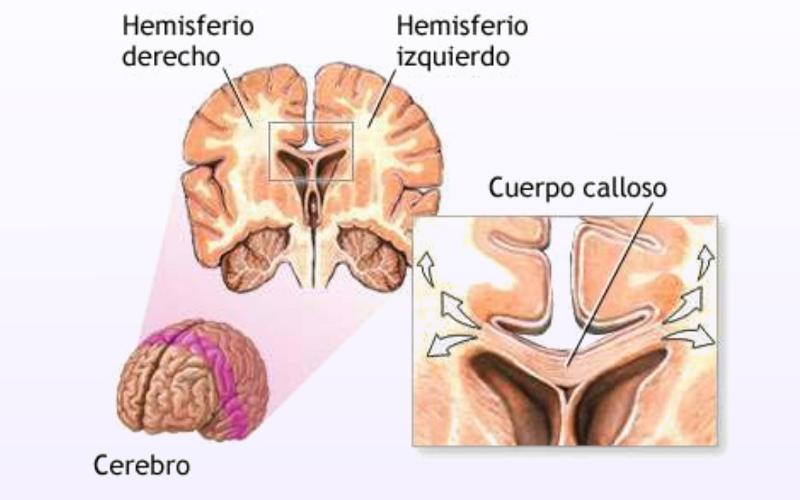 Este síndrome puede suceder en pacientes con infecciones, tumores, golpes o cirugías cerebrales que afectan a-- las comisuras que unen ambos hemisferios cerebrales (Cuerpo calloso, fórnix, comisura blanca). Adam Así pues, este es un trastorno neurológico y aunque la descripción suena extraña, los síntomas en los pacientes siempre coinciden. El paciente con el síndrome de la mano extraña aunque puede sentir tacto en la mano, cree que no es parte de su propio cuerpo y no posee control sobre sus movimientos. Las manos aquejadas pueden realizar actos complejos pero muchas veces el paciente no es consciente de lo que su mano extraña hace, hasta algún acto de esta llama su atención. Frecuentemente personifican el miembro independiente, pensando si tienen creencias religiosas que se encuentran "poseídos" por algún espíritu y llega a enfrentarse a su propia mano o castigarla para intentar controlarla. Solo en casos muy extremos, la mano termina significando un peligro real para el paciente al provocarse autolesiones. Ciertamente, a día de hoy no existe ningún tratamiento conocido para el síndrome de la mano extraña, aunque se considera que este síndrome resulta de la desconexión entre las distintas partes del cerebro con control sobre el cuerpo y se ve incrementado en situaciones de fatiga, ansiedad u otras donde el paciente se encuentra sin prestar atención a su miembro. Con todo, los síntomas pueden ser disminuidos ocupando la mano extraña con alguna tarea que permita mantener un control activo sobre ella o reduciendo el estrés del paciente, resultando en poco más que una molestia con el trabajo profesional adecuado en muchos casos. Síndrome de Rapunzel Otra dolencia de carácter mental muy conocida y desafortunadamente confundida con una enfermedad rara es este trastorno psicológico compulsivo: tricofagia, es decir, el acto de comer pelo de forma compulsiva, que además en algunas ocasiones, se asocia con la tricotilomanía, un hábito recurrente e irresistible dirigido a arrancarse el propio cabello o los vellos de distintas zonas del cuerpo. A la larga esta condición intestinal provoca una compleja afección con dolor y tensión abdominal, la pérdida del apetito, náuseas, vómitos, pérdida de peso, sangrado y perforación intestinal. Como su nombre indica, recibió este nombre por la joven de la larga cabellera Rapunzel, de la clásica historia de los hermanos Grimm, pero ni la tricofagia ni la tricotilomanía por mucho que se suelan mencionar en las listas de enfermedades raras, lo son en sí. Hablamos de una condición intestinal extremadamente rara, pero peligrosa, eso sí, ya que el tracto gastrointestinal humano es incapaz de digerir el cabello, y el tricobezoar (acumulación de pelo no digerible, capaz de formar un masa de volumen considerable), debe ser removido quirúrgicamente. 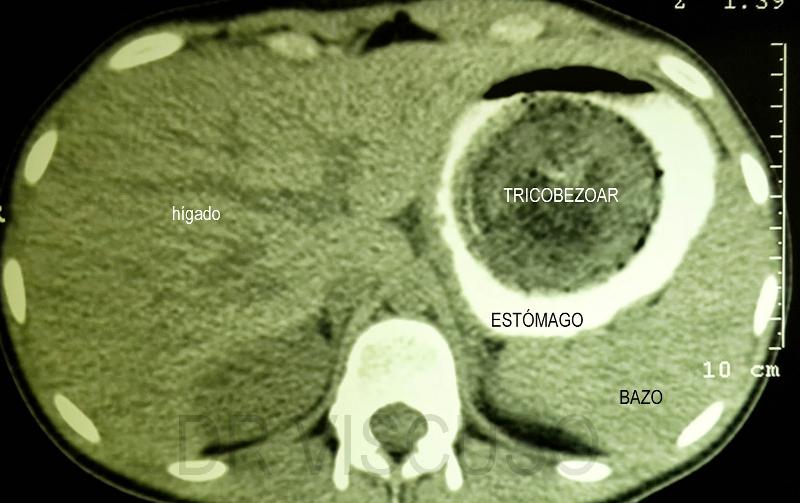 Tomografía una de niña de 14 años, con alteraciones psiquiátricas, que se presenta con una severa pérdida de peso, anemia, y alteraciones digestivas inespecíficas. Ante lo poco característico del cuadro se solicita una tomografía computada de abdomen donde se visualiza una infrecuente masa ocupante de la cavidad gástrica. Se confirmo el diagnóstico por gastroscopia, de tricobezóar (masa de cabellos ingerida por la paciente durante muchos meses, y se intentó la fragmentación y extracción endoscópica. Esto no se logró por lo compacto de la masa de pelos. Es operada extrayéndose un gran molde del estómago formado por cabello. Antonino En estos casos, los pacientes requieren de una evaluación y tratamiento psiquiátrico debido a su asociación con trastornos del control de impulso, como puede ser la cleptomanía, la piromanía, la ludopatía o la compra impulsiva, y aunque es un grupo de trastornos de etiología desconocida, están muy estudiados en la medicina psiquiátrica existiendo unas características comunes, ya que el sujeto no puede resistirse al impulso o tentación de llevar a cabo dichas acciones, aunque sean peligrosas. De igual manera antes de llevar a cabo la acción de su trastorno hay un aumento de la tensión emocional y durante la acción concreta se experimenta una sensación placentera o liberadora que puede acabar más tarde en arrepentimiento o culpabilidad, como sucede comúnmente en la ludopatía o las compras compulsivas, así que una vez más, incluir el síndrome de Rapunzel entre las enfermedades más raras resulta un error. Ilusión de Cotard o síndrome del muerto viviente (CIE-9 297.1) Muchas veces este síndrome, también conocido como delirio de negación, delirio nihilista y más recientemente como síndrome zombie o del muerto viviente en artículos de prensa o internet, se le califica de forma completamente errónea como la enfermedad más rara del mundo. Una vez más su etimología viene de lejos: este síndrome recibe su nombre de Jules Cotard, neurólogo francés el cual descubrió este síndrome, llamándolo inicialmente "delirio de negación" cuando lo expuso por primera vez en el año 1880. Cotard lo describió, explicando el caso de un paciente, el cual negaba la existencia de Dios y el diablo, así como de diversas partes de su cuerpo y de la necesidad de alimentarse. Con el paso del tiempo y el agravamiento de su delirio, creyó estar eternamente condenado y que no podría morir, debiendo vagar eternamente por la tierra si no se le daba muerte de alguna manera específica. Desde entonces se han ido contabilizando diversos casos, y es un delirio típico de las depresiones más graves, pudiéndose encontrar en otras enfermedades mentales severas, como demencias con síntomas psicóticos, esquizofrenia, psicosis debidas a enfermedades médicas o a tóxicos. Comúnmente consta de un avanzado desorden mental hipocondríaco tras el cual el paciente cree estar muerto, física y literalmente, con sus órganos en putrefacción, pero por alguna razón siguen conscientes entre los vivos, (muchas veces, esa razón a la que culpan de su estado, es de tipo religioso y asociada a maldiciones y demonios). A veces en su delirio, el paciente se mira en el espejo y no logra reconocer su rostro, viendo un cadáver en su reflejo y llega a creer que nunca podrá morir realmente y que será finalmente un zombie para el resto de la eternidad.  psiquiatria.com Ya en 1996, hace ya más de 20 años. Young, A.W. & Leafhead, K.M. escribieron: Betwixt Life and Death: Case Studies of the Cotard Delusion, donde describen con precisión uno de estos casos, así como los distintos mecanismos que usa el delirio para nutrirse: Los síntomas del paciente se dieron en el contexto de sensaciones más generales de irrealidad y de estar muerto. En enero de 1990, después de recibir el alta en el hospital de Edimburgo, su madre lo llevó a Sudáfrica. Estaba convencido de que había sido llevado al infierno (lo que se confirmaba por el calor), y que había muerto de septicemia (que había sido un riesgo al principio de su recuperación), o quizá de sida (había leído una historia en The Scotsman acerca de alguien aquejado de sida que había muerto de septicemia), o de una sobredosis de una inyección contra la fiebre amarilla. Pensaba que se habían «apropiado del espíritu de mi madre para mostrarme el infierno», y que seguía dormido en Escocia. Una vez más, pese a lo extraño del comportamiento del paciente, no deja de ser un delirio que puede aparecer asociado en el contexto de una enfermedad neurológica o mental, estando perfectamente reconocido y resultando por tanto tratable con las herramientas disponibles en neuro psiquiatría. Síndrome de la cabeza explosiva Este síndrome, documentado por primera vez por el médico Silas Weir en el 1876 cuando se refirió a dos hombres que sufrieron lo que llamó "descargas sensoriales", es descrito por el paciente como un sonido extremadamente fuerte, que generalmente describe como un estallido o explosión, que parece venir del interior de su propia cabeza. Pero, aunque se percibe como un sonido increíblemente fuerte, muy rara vez viene acompañado de dolor, aunque también se han documentado casos en ocasiones de flashes luminosos o dificultad para respirar. De todas maneras la casuística es muy variada: algunos la describen como una bomba que explota al lado de sus cabezas cuando caen dormidos y solo les sucede una vez en la vida, mientras que para otros pacientes puede llegar a suceder en muchas ocasiones en una misma noche y de manera continuada. Si bien este síndrome ni siquiera posee código CIE, es real y está más ampliamente documentado de lo que se piensa, pero hay que aclarar las cosas: A día de hoy existe una teoría de que esta condición está relacionada estrechamente con las alteraciones del sueño y además podría ayudar a explicar fenómenos culturales aparentemente no relacionados y con conexiones con la psiquiatría, como las creencias de fantasías y delirios como los secuestros alienígenas, teorías de conspiraciones gubernamentales o posesiones. El profesor adjunto de psicología de la Universidad de Washington State, Brian Sharpless, encabezó un estudio cuyos resultados se publicaron en mayo del año 2015, donde se le preguntó a 211 estudiantes si habían tenido esta experiencia y sorprendentemente 18% respondió afirmativamente.  Silas Weir Mitchell (1829 –1914) Este médico americano no solo es conocido por describir por primera vez el síndrome de la cabeza explosiva, sino también por el descubrimiento de la Causalgia (Síndrome de dolor regional complejo) y la Eritromelalgia (enfermedad vascular periférica rara que se manifiesta por crisis repetidas de hipertermia y dolor en manos y pies, que se desencadenan por el calor). Varias teorías han tratado de explicar su origen con el paso de los años, incluyendo trastornos del oído y convulsiones epilépticas parciales, aunque la teoría más sugestiva procede de algunos estudios realizados a individuos a quienes se les monitorizó la actividad cerebral mientras dormían. Los estudios sugieren que quizás, pueda existir un estallido de actividad neuronal en el cerebro que coincide con el momento de citada explosión. Normalmente cuando vamos a dormir, el cerebro normalmente se va desactivando poco a poco, pero quizás, en el síndrome de la cabeza explosiva, sobre todo en pacientes con problemas de sueño (recordemos el estudio, pues los estudiantes son propensos a la falta de sueño) existe una interrupción en la formación reticular, la parte del cerebro responsable de supervisar esa desconexión progresiva, que tiene como consecuencia un atraso en desconectar algunas áreas. Esa demora está asociada con una supresión de las ondas del cerebro alfa normalmente responsables de la somnolencia y un súbito estallido de actividad neuronal en las zonas del cerebro responsables por el procesamiento del sonido, sintiendo que todas las neuronas se están disparando al mismo tiempo", provocando la sensación de una explosión en la cabeza. Poco más podemos decir a día de hoy sobre el síndrome de la cabeza explosiva, sin consecuencias más allá de un susto si no está ligado a otros síntomas (y solucionable fácilmente con una vida sin estrés y dormir adecuadamente). Curiosamente el síndrome de la cabeza explosiva está muchas veces ligado a la parálisis del sueño, otro desorden del sueño en el que uno se siente como si estuviese despierto, pero sin poder mover el cuerpo y que más de un lector habrá experimentado probablemente sin mayores consecuencias que un angustioso recuerdo. Brian Sharpless considera que ambas experiencias podrían explicar algunos relatos aparentemente sobrenaturales ya que tanto la parálisis del sueño como el síndrome de la cabeza explosiva parecen compartir un problema subyacente en la transición entre estar despiertos y dormidos, que con determinados condicionamientos sociales o culturales podrían llevar al paciente a creer que ha sido abducido o bien poseído por un demonio. Pero a día de hoy solo son hipótesis que hay que seguir estudiando. Macropsia y Micropsia (Síndrome de Alicia en el País de las Maravillas) Aquí tenemos de nuevo dos relativamente extraños trastornos neurológicos frecuentemente confundidos con ERs. La Micropsia, también conocida como el Síndrome de Alicia en el país de las maravillas, resulta en un trastorno de la percepción donde los pacientes describen episodios en los que pueden ver los objetos más grandes o más pequeños, alejados o cercanos, incluso pueden tener la sensación de que una parte de su cuerpo es muy grande o muy pequeña. Estas alteraciones, tanto ver los objetos excesivamente grandes (Macropsia) como pequeños (Micropsia), pueden afectar también a la apreciación del tiempo haciéndolos sentir que el tiempo ha pasado muy lento o muy rápido. Los síntomas pueden durar pocos minutos a algunas horas y pueden ocurrir varias veces al día o una vez al mes o cada varios meses. Estos síndromes se asocian en la mayoría de los casos al desarrollo posterior de una crisis de migraña o también puede constituir la antesala a una crisis epiléptica. Muchas veces se confunde con una enfermedad, cuando realmente los síntomas de este síndrome pueden ser debidos a múltiples razones tanto a orgánicas como psicológicas. Puede presentarse como un síntoma asociado a una crisis de migraña o epiléptica, pero también por causas tan dispares como por alteraciones de la retina como ocurre en el caso del desprendimiento de esta capa profunda del ojo o en trastornos neurológicos con daño a las vías nerviosas relacionadas con la visión que puede ocurrir a consecuencia de un accidente cerebrovascular, hemorragia cerebral, una lesión ó tumor cerebral bien sea benigna o maligna. 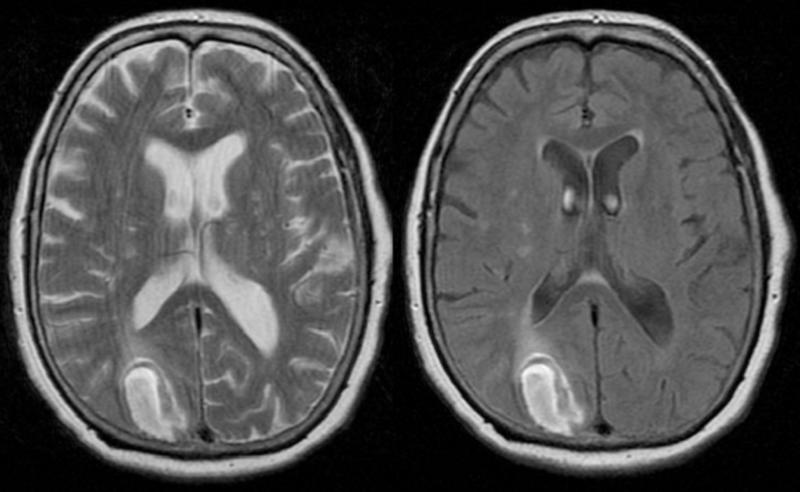 Síndrome de Alicia en el país de las maravillas en patología cerebrovascular. RMN cerebral. Lesión hiperintensa en T2 y FLAIR en su periferia relacionada con hemorragia intraparenquimatosa de distribución corticosubcortical occipital derecha. Durante su ingreso, el paciente presentó episodios transitorios y repetidos de ilusiones ópticas, tales como notar que en el hemicampo visual izquierdo las imágenes se distorsionaban (dismorfopsia), «veía la figura de mi esposa alargarse el cuerpo a la vez que se alejaba» (macropsia y teleopsia respectivamente), «veía gente en el lado izquierdo que estaban vestidos de blanco, veía coches que cruzaban de un lado a otro y siempre eran de color blanco», «mi esposa estaba de blanco y cuando se ponía a mi derecha aparecía con la ropa que realmente llevaba puesta», y notaba aparición de figuras geométricas e inversión de la imagen en dicho campo visual, «las imágenes iban como lentas y a veces parecían como si se congelaran» (palinopsia). Por el hallazgo en electroencefalograma y la sintomatología indicativa de crisis focal del lóbulo occipital, se inició mediación antiepiléptica con levetiracetam, mejorando considerablemente la sintomatología, llegando a desaparecer por completo. Elsevir- Revista de Neurología. J.L. Camacho Velasquez También se ha descrito la percepción de objetos más grandes de lo que representan en pacientes con una infección por el Virus Epstein Barr, causante de la Mononucleosis infecciosa o el Virus Coxsakie. Existen algunas causas psicológicas que pueden explicar la aparición de macropsia como son algunos estados de ansiedad, miedo, angustia y trastornos psiquiátricos acompañados por disociación con la realidad. Los síntomas son increíblemente variados dentro de la alteración, dependiendo de las distintas causas que lo originan, podemos describir por ejemplo la micropsiapsia de convergencia-acomodativa, donde los objetos se vuelven más pequeños a medida que la persona se acerca, la micropsia psicógena que se presenta en personas con ciertos trastornos psiquiátricos más complejos, micropsia de retina, donde se aumenta la distancia entre los fotorreceptores de la retina y se disminuye la agudeza visual y la micropsia cerebral que acostumbra a presentarse en niños con migrañas crónicas. En los casos contrarios, donde las personas ven los objetos más grandes de lo que son en realidad, hace que estas personas se sientan más pequeñas por comparación, aunque la mayoría de los pacientes son conscientes de que lo que están viendo no se corresponde con la realidad pero les provoca graves dificultades para situar un objeto en el espacio, apreciar la simetría o determinar el tamaño real del objeto. La macropsia principalmente ha sido descrita como un efecto secundario del uso de drogas ilícitas con poder alucinógeno o por la administración de algunos medicamentos como el topiramato empleado para la prevención de las migrañas. el zolpidem empleado para el tratamiento del insomnio o el citalopram empleado como antidepresivo. En resumen, hablando con propiedad estos síndromes de la apreciación del espacio están lejos de ser considerados una ER y sí la consecuencia sintomática de múltiples dolencias tanto físicas como psíquicas o la administración de ciertos fármacos o sustancias alucinógenas, que producen esta curiosa alteración perceptiva. Síndrome de Capgras Uno de los últimos ejemplos de condiciones mentales que erróneamente se confunden con enfermedades raras que vamos a analizar en este reportaje es un viejo conocido, comúnmente confundido y ejemplo paradigmático de lo que realmente no debe ser descrito como una enfermedad de este tipo. Este síndrome afecta el sentido de la identificación del paciente. Se caracteriza por la constante creencia de que un ser cercano y querido fue sustituido o reemplazado por un impostor idéntico. De hecho, esta condición no es en realidad una enfermedad paranoide específica y ni tan siquiera aparece como tal en el CIE (clasificación internacional de enfermedades) ni en el DSM (manual diagnóstico y estadístico de los trastornos mentales), así se describe separadamente por manifestar ciertas características sintomáticas que pueden aparecer a manera de fenómeno único o acompañar incluso a varios tipos de psicosis. Para variar, otra vez, es ya un antiguo conocido de la psiquiatría, fue descrito por primera vez hace ya casi un siglo en el año 1923, bajo el nombre de l'illusiondes sosies (ilusión de los dobles) por Jean Marie Joseph Capgras, al informar sobre un caso de delirio en el que otras personas normalmente muy cercanas al paciente habían sido reemplazadas por dobles exactos. El impostor, adquiere para el paciente los mismos rasgos físicos que la persona “original”, pero su mente pertenece a ella. Esta distinción es crucial, pues evidencia la creencia por parte del paciente de creer que hay dos personas físicamente iguales, pero distintas en aspectos mentales. El paciente que padece síndrome de Capgras, lo lleva a imaginar que el impostor actúa de la misma manera que el sujeto original para confundirlo y engañarlo con el objeto de que piense que se trata del individuo original, muchas veces ocultando oscuras intenciones. Hace casi un siglo Joseph Capgras explicó el delirio como “el resultado del sentimiento de extrañeza, combinado con una tendencia paranoide a desconfiar, así como una ambivalencia del paciente con su entorno más cercano”. Pero hoy en día y tras un siglo estudiando patologías más comunes, el síndrome de Capgras permanece eso sí como uno de los casos menos frecuentes dentro del mundo de la psiquiatría, aunque se ha informado de numerosos casos y variantes que también se han relacionado con otros síndromes. 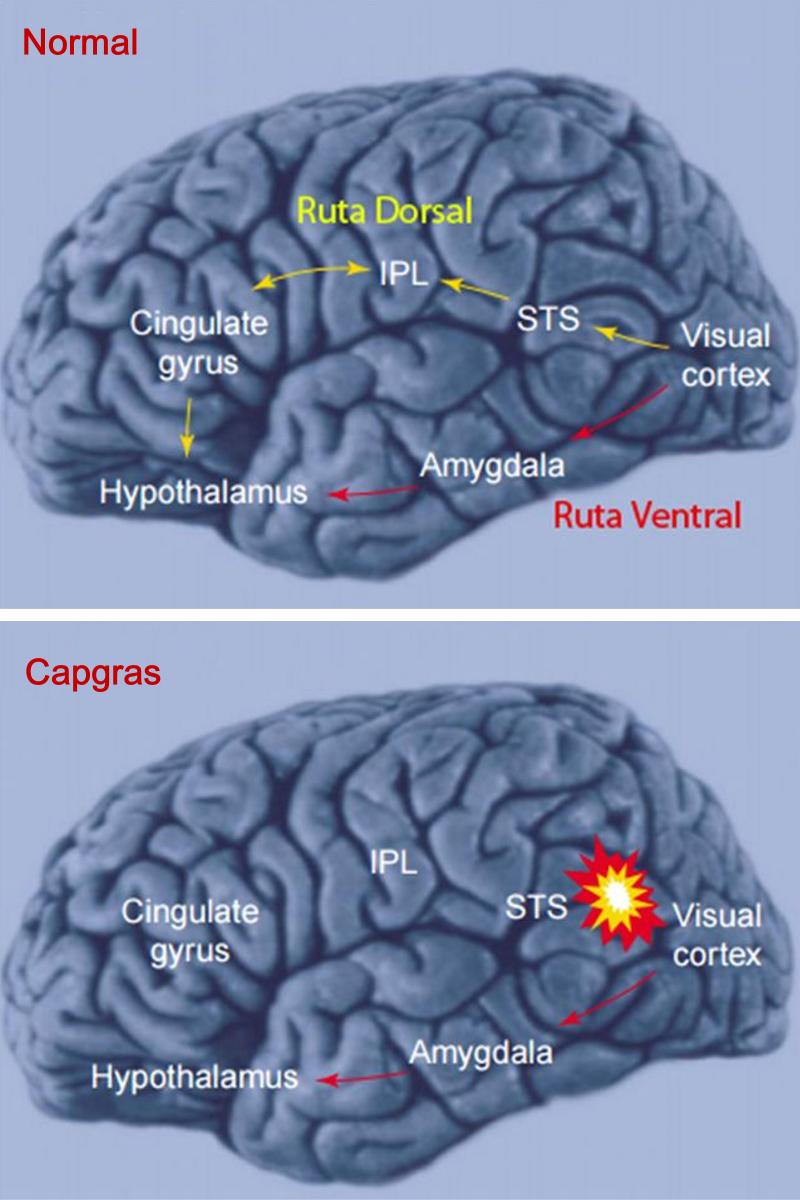 Neuroanatomía del procesamiento facial. La ruta amarilla muestra la ruta inconsciente dorsal la cual pasa por el lóbulo parietal inferior (IPL) y el surco temporal superior (STS). La ruta roja es la ruta consciente de reconocimiento ventral. En Capgras, el daño se conjetura que ha ocurrido en la ruta dorsal inconsciente. Naukas / Eva Vico Así, algunos de los primeros indicios de las posibles causas del síndrome de Capgras se encontraron tras el estudio de pacientes con lesiones cerebrales que habían desarrollado la Prosopagnogsia (Interrupción selectiva de la percepción de rostros, tanto del propio como el de los demás, que pueden ser vistos pero no reconocidos como los que son propios de una determinada persona). Al parecer los delirios se producen con más frecuencia tras lesiones en el hemisferio derecho y en la zona bifrontal, porque afectan funciones como la auto monitorización, la monitorización de la realidad, la memoria y la sensación de familiaridad, considerando, además, la necesaria preservación del hemisferio izquierdo para su aparición. Así pues entre las teorías para explicar este delirio se han propuesto distintos síndromes de desconexión entre las diversas estructuras cerebrales implicadas, así como otras explicaciones sobre la laterización y la localización de las disfunciones que originan el delirio. El síndrome es de difícil tratamiento debido a que se trata normalmente de pacientes con lesiones cerebrales que han desarrollado la Prosopagnogsia, pero a día de hoy se aplican medicamentos anti-psicóticos, anti-depresivos, así como terapias cognitivas y de conducta con cierto éxito, aunque ninguna asegura una cura con certeza y se deben estudiar los casos de manera individual para elegir el mejor tratamiento. Por tanto a día de hoy, dentro de la rama de la psiquiatría el síndrome de Capgras está incluido dentro de un subgrupo de patologías conocidas como Síndrome de Falta Identificación Delirante (SFID), donde se encuentran también el síndrome de Frégoli, (un trastorno psíquico de tipo delirante y paranoico que se caracteriza por la creencia de que las personas en realidad son siempre un único sujeto que cambia su apariencia o se disfraza para parecer diferentes individuos). síndrome de Intermorforsis o el síndrome de dobles subjetivos. Todos ellos implican procesos complejos, que no se limitan a un simple problema de procesamiento facial, sino a una disfunción múltiple basada en procesos cognitivos implicados en la interpretación de percepciones anómalas y en la formación de creencias que dificultan el tratamiento, agravado la mayor parte de los casos por daños cerebrales permanentes como la Prosopagnosia que ya hemos citado, pero que se han estudiado y se estudian desde hace años en diversos campos de la medicina, obteniéndose continuamente resultados positivos y progresos claros a medida vamos conociendo mejor la mecánica de nuestra mente. Sería por tanto este, el ejemplo perfecto de un síndrome comúnmente relacionado erróneamente con las enfermedades raras en los medios de comunicación, pues no cumple ninguno de los requisitos: es largamente reconocida y descrita, se conocen las causas y los síntomas perfectamente, lo que permite un diagnóstico claro, está asociada a otros problemas de origen neurológico y a día de hoy la medicina, tras años de estudio ni siquiera la reconoce como una enfermedad propiamente dicha. Y la lista de confusiones podría seguir y seguir… Una vez aclarado con ejemplos lo que no es propiamente una ER y antes de entrar en las ER propiamente dichas, vamos a mencionar someramente otras “dolencias” (por englobarlas de algún modo), que frecuentemente se incluyen por cuestiones sensacionalistas o morbosas en las listas de enfermedades raras sin demasiado criterio: Síndrome del marido jubilado Entre todas las enfermedades raras mal entendidas, sin duda esta se lleva un premio especial, pues para ser mencionada como poco frecuente se estima que hasta el 60% de la población japonesa femenina de la tercera edad padece este síndrome. Lo más curioso es que existir, existir… si existe y fue reconocido oficialmente en el año 1991, en Japón, aunque se trata simplemente de una dolencia psicosomática, vinculada con el estrés y la depresión, donde una mujer progresivamente muestra signos de una enfermedad física y cuadros depresivos cuando su marido llega o se aproxima a la jubilación (mejor nos abstenemos de comentarios particulares del papel de la sociedad en este síndrome). Síndrome de París (anti Stendhal) La idiosincrasia de la cultura Japonesa es realmente muy especial, teniendo en este síndrome malentendido otro de sus principales exponentes, pues se trata de un trastorno psicológico momentáneo y poco grave realmente, que se ha documentado casi únicamente con turistas de dicha nacionalidad. Se produce obviamente en turistas que visitan la capital francesa y la crisis se produce cuando los visitantes llegan a la ciudad, comienzan la visita y sus atractivos artísticos no coinciden con lo que hasta entonces eran sus expectativas, momento a partir del cual experimentan una especie de síndrome de Stendhal pero por razones y consecuencias casi inversas, presentando cuadros depresivos, mucha ansiedad y síntomas, como ideas paranoides y una desilusión tan aguda que les puede llevar en ocasiones a considerar que son víctimas de maltratos y agresiones por parte de los parisinos.  Sin duda la moderna idiosincrasia social japonesa es capaz de exportar no solo su propio estilo de comics y videojuegos, si no sus propios síndromes particulares. Síndrome de Stendhal Obviamente y tras mencionar su alter ego, tenemos que mencionar este trastorno psicosomático, también conocido como síndrome de Florencia o estrés del viajero, que es probablemente el síndrome menos raro o desconocido que podamos imaginar (empleado incluso como recurso para un célebre anuncio publicitario). Los síntomas en cuestión se manifiestan psicosomáticamente al ver obras de arte, sobre todo si se trata de una obra muy bella o de varias obras de excepcional calidad emplazadas en un mismo lugar, que provocan en la persona afectada un intenso vértigo, confusión, ansiedad, aceleración del ritmo cardíaco, temblores, palpitaciones, depresión e incluso alucinaciones. Glosodinia o el síndrome de la boca ardiente Una ¿enfermedad? Con un origen aún desconocido. Los pacientes afirman sufrir una intensa sensación de ardor y quemazón en la lengua que persiste por largo tiempo. Es cierto que se han registrado numerosos casos alrededor del mundo, pero no existen análisis que hayan detectado anomalías en la lengua o a nivel de daños cerebrales a día de hoy. Esta enfermedad, de serlo, es todo un misterio y no provoca daños de otro tipo o empeoramiento de la salud del paciente. Trimetilaminuria o el síndrome del olor a pescado Conocido desorden metabólico que provoca un fallo en la producción de la enzima flavinmonooxigenasa 3 (FMO3) y que tiene por consecuencia un olor a pescado muy intenso que se expele en el aliento, la orina y la transpiración. Es real y aún no existe cura ni tratamiento, es más común de lo que se piensa, con muchos caso documentados, pero tan solo se limita a esta desagradable característica, sin poner en ningún caso en peligro la vida o la integridad del paciente (más allá de los efectos psicológicos de rechazo que puede provocar, sin una higiene exquisita y concienzuda). Síncope hilarante o desmayo por risa Este caso no deja de ser una forma poco usual de síncope situacional, donde los afectados por esta condición se desmayan cuando se ríen demasiado y aunque muy poco se sabe al respecto, hay varios casos documentados de este, como mucho ¿síndrome? que se cree tiene un origen fisiopatológico. Disgeusia Provoca en los afectados un cambio en la percepción de los sabores de bebidas y alimentos, muchas veces provocando una sensación muy desagradable, transformando un sabor que puede considerarse agradable en uno repulsivo. Es un trastorno poco conocido todavía, aunque existen bastantes casos documentados y sus causas parecen estar relacionadas con otros trastornos neurológicos poco comunes, pero normalmente estos síntomas no provocan mayores problemas a no ser que los fallos neurológicos asociados conlleven otros síntomas y consecuencias para el paciente. Delirio “The Truman Show” Otro curioso delirio persecutorio y de grandeza que se enmarca sin demasiado criterio dentro de las ER. Como su nombre indica procede del clásico film dirigido por Peter Weir y protagonizado por Jim Carrey, donde la vida del personaje principal se desarrolla retransmitida en un programa de televisión con formato de reality show sin su conocimiento. Este delirio realmente ha adoptado varios nombres a lo largo del tiempo y quienes se supone lo padecen están convencidos de ser la estrella de un reality show imaginario las 24 horas de cada día, por lo que no deja de ser un problema clásico de la psiquiatría adaptado a una de las miles de combinaciones del contexto socio cultural moderno.  A medida evoluciona nuestra sociedad, de igual manera lo hacen sus síndromes, en la Edad Media y aún hoy en día en los lugares más religiosos, los pacientes (o las víctimas en algunos casos) veían aparecerse al Diablo por todas partes… ¿Hemos sustituido al diablo por la celebridad de la cámara y las redes? wallpapertag Síndrome del acento extranjero Otra condición consecuencia normalmente de una lesión cerebral grave o un accidente cerebrovascular. Quienes padecen la condición desarrollan lo que parecería ser un acento extranjero como efecto secundario de la lesión, consecuencia de fallos en la articulación y los procesos de coordinación, causados por los problemas neurológicos de la lesión cerebral. Una vez más se elimina el contexto para enmarcar estos síntomas como una enfermedad propiamente dicha. Discronometría Otro ejemplo, como el anterior, donde se confunde la “parte” con el “todo” tras las consecuencias de un daño cerebral (Esta concretamente sucede cuando se daña el cerebelo y pierde parte de su funcionalidad.) Quienes se ven afectados no pueden calcular con precisión la cantidad de tiempo que va pasando, sufriendo una distorsión en su percepción. Convulsiones por escuchar a Mary Hart Para terminar la lista de errores y confusiones, sin duda podemos decir que la más hilarante que hemos llegado a ver en Internet sin duda es esta. Aunque curiosamente se basa en un caso real documentado, Mary Hart, veterana estrella de la televisión estadounidense, protagonizó involuntariamente este caso concreto. La revista médica New England Journal of Medicine, publicó en 1991, un artículo donde documentaba un curioso caso de epilepsia, donde las convulsiones parecían haber sido provocadas en el paciente aquejado de dicha enfermedad por la voz de Hart en una emisión de su show Entertainment Tonight. Los médicos consideraron los ataques de epilepsia causados por una determinada frecuencia con el nivel y el tono de voz exacto de la presentadora en la fatídica noche. A día de hoy, las advertencias en la exposición a los videojuegos por parte de pacientes con epilepsia o en conciertos, por ejemplo son normales sin hablar por ello de la enfermedad de “Call of Duty” o “Super Mario”… todo sea por un buen titular. Reflexionando en serio:Neuropatía hereditaria sensitivo autonómica o insensibilidad congénita al dolor. (CIE-10: G60.8) Una vez abandonamos el campo de la psiquiatría y los síndromes derivados de trastornos neurológicos, una de las primeras enfermedades que suele destacar por su extraña condición es sin duda esta. Resulta muy conocida por el público en general y básicamente es una condición médica en la cual la persona nace con la incapacidad de sentir dolor, (aunque deberíamos hablar realmente no solo de dolor, sino también de tacto y sensibilidad en distintos niveles, según el tipo) pero su organismo es completamente funcional, con ciertas excepciones como en el tipo IV donde existe además una deficiente regulación de la temperatura corporal y es frecuente el fallecimiento por aumento de temperatura corporal (hiperpirexia), así como habitualmente la existencia de retraso en el desarrollo mental. Lógicamente, esto acarrea graves problemas para el paciente, ya que es el dolor lo que nos advierte del peligro y nos ayuda a mantener nuestra seguridad e integridad física, pero en los individuos con insensibilidad congénita al dolor no existe reacción de huida ante estímulos dolorosos La Neuropatía hereditaria sensitivo autonómica, (realmente es un conjunto de enfermedades hereditarias poco frecuente) pero por suerte hay bastante información de esta enfermedad, pese a su rareza: se debe a una mutación genética en la síntesis de un tipo de canal de sodio en particular, el cual se encuentra en el sistema nervioso y en las neuronas, encargándose de transmitir, enviar y recibir el dolor en el sistema nervioso central.  Por mucho que nos quejemos y maldigamos, el dolor es un mecanismo básico de supervivencia, su ausencia total no es una suerte en ningún caso. La lucha médica consiste en “desactivarlo” una vez que las alarmas han comenzado y ya son innecesarias. postimage En los pocos casos que se han registrado de esta mutación, los pacientes han fallecido muy jóvenes por infecciones, ulceraciones y otras condiciones que se detectado demasiado tarde por la falta de alarma o incluso hasta por heridas causadas por ellos mismos. En un principio la enfermedad y sus distintos tipos, no es mortal por si misma (excepto en casos concretos como el tipo IV que ya hemos mencionado) pero la ausencia del mecanismo de alarma más básico en nuestro cuerpo y del cual muchas veces nos quejamos: el dolor, puede causar terribles consecuencias si desaparece. Se calcula que existen unos 100 casos documentados en Estados Unidos y más de 300 en Japón debido a que la enfermedad es más propicia en sociedades genéticamente homogéneas, pero se desconoce la frecuencia en otras áreas del mundo, pues muchas veces no es diagnosticada al pasar desapercibida para la medicina o por cuestiones de precariedad sanitaria. Podemos decir sin equivocarnos que muchas veces el dolor nos mantiene vivos. Urticaria (Prurito) acuagénico (CIE-10 L29.8 <ILDS L29.83>) Dentro de las ER propiamente dichas, existe todo el espectro de gravedad como en las enfermedades habituales, entre las menos graves y conocidas encontramos la citada urticaria, que pese a lo sorprendente que resulte ser “alérgico al agua” pues es una de las bases de la vida, resulta ser una enfermedad documentada. Si bien no tiene consecuencias fatales, la intensidad del picor o la irritación puede llegar a impedir el desarrollo las actividades normales al paciente, como pueden ser problemas de higiene y aseo que conducen muy frecuentemente a la depresión severa, por lo que después de recibir un diagnóstico afirmativo, muchos pacientes expresan un gran alivio, dada la incredulidad con la que su enfermedad es tomada en muchos casos. Pero ciertamente es una de las formas de urticaria física, en la que las lesiones en la piel son consecuencia de estímulos o cambios físicos del medio ambiente. La urticaria acuagénica es más común en las mujeres y los síntomas a menudo comienzan en torno a la aparición de la pubertad. 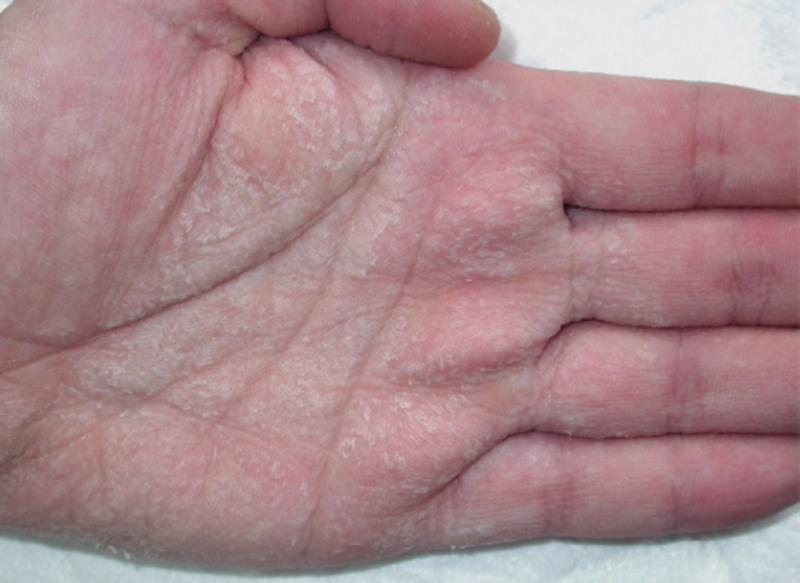 Caso clínico de urticaria acuagénica en un varón de 28 años, que se desencadenaban a los pocos minutos de sumergir las manos en agua y remitían al cabo de una hora de secarlas, presentaba placas blanquecinas, aterciopeladas, con aspecto de «empedrado» en la palma y los dedos. En la superficie de estas placas se advertían numerosos orificios puntiformes. Las lesiones eran más intensas cuanto mayor era la temperatura del agua y el tiempo de exposición. Actasdermo / M.A. Pastor / Servicio de Dermatología. Hospital Santa Bárbara. Puertollano. Ciudad Real. España La causa exacta no se sabe todavía. Debido a la rareza de la condición, hay muy pocos datos sobre la eficacia de los tratamientos individuales, sin embargo, varios medicamentos y terapias se han utilizado con cierto éxito en casos determinados. Realmente los científicos no creen que la molécula en si del agua sea la responsable del problema, se considera más bien dos teorías:
A día de hoy es un misterio para la ciencia, aunque las consecuencias físicas han permitido documentarla y estudiarla, lo cual, unido a lo espectacular del principio causante y la facilidad para su explicación ha facilitado que también sea ampliamente difundida en los medios de comunicación, aunque esta vez si hablamos de una ER propiamente dicha, pese a que no queda claro si realmente las causas son externas, independientemente del paciente, lo cual se fundamenta en que la mayoría de los casos de urticaria acuagénica son esporádicos y ocurren sin que haya otros casos en la misma familia, donde raramente, más de una persona se ve afectada con esta condición. Hipertricosis (CIE-1 L68, Q84.2) Enfermedad del hombre lobo. Mencionada en todas las listas espectaculares de enfermedades raras, esta vez sí que tenemos que aceptarla por derecho propio dentro de lo que sería por definición una autentica ER y no solo porque sus síntomas exteriores sean claros y evidentes. Al fin y al cabo la esperanza de vida teórica de estas personas es la misma que la de una persona normal, con la única diferencia que del pelo. Las personas que la padecen están cubiertas completamente con la excepción de las palmas de las manos y de los pies, si bien no es el pelo normal, como el de la cabeza sino lanugo un pelo blanquecino y fino, parecido a la pelusa, que aparece en los recién nacidos pero que desaparece tras los primeros meses de vida pero que sin embargo en la hipertricosis continúa y crece durante toda su vida. Se debe a una mutación genética adquirida normalmente por herencia familiar, pudiendo darse en varios miembros, además se conocen dos variantes: La hipertricosis universal congénita, autosómica dominante y la hipertricosis generalizada congénita: autosómica dominante o recesivo y ligado al cromosoma X. Por suerte afecta tan solo a 1 de cada 1000 millones de personas y desde la Edad Media hasta nuestros días tan solo se han registrado 50 casos aproximadamente.  Este es un caso real donde por desgraciada la hipertricosis se ve agravada por el síndrome de Byars-Jurkiewicz. Se trata de una niña de 12 años, llamada Akhtar, originaria de Bangladesh. Para empeorar su situación el síndrome de Byars-Jurkiewicz, crea desórdenes faciogenitales masivos y un desarrollo exagerado de los pechos tras la pubertad. A causa del síndrome, las encías de Akhtar se han hinchado hasta recubrir sus dientes y sus pechos han crecido exageradamente, hasta totalizar casi la mitad de sus 38 kilos de peso, lo que le impide permanecer de pie o ir a la escuela. Ahora bien pocas enfermedades en la historia, son tan relevantes para mostrar lo que es en el ser humano la lacra del miedo al diferente, la sociedad, en su ignorancia hace que la persona que tiene esta enfermedad se vea aislada, sea discriminada y en ocasiones, maltratada física y psicológicamente. Junto con la Porfiria Cutánea Tarda, enfermedad donde se acumulan Porfirinas, las cuales en contacto con los rayos ultravioleta del sol generan excitación en las moléculas, liberando radicales libres, dañinos para los tejidos y la piel, provocando que la piel se defienda aumentando la cantidad de pelo en las zonas en contacto solar para así evitar el daño. Las personas con esta enfermedad, durante siglos no diagnosticada, ni conocida solían salir por la noche para evitar el sol, a ser posible en noches de luna llena para poder ver mejor bajo su luz. De tal modo la ignorancia y el miedo contribuyeron a crear el mito del hombre lobo al amparo de estas dos enfermedades no conocidas durante siglos y que marcaron la desgracia de sus portadores. Junto con el Albinismo (CIE-10 E70.3) que tan solo se trata de una alteración genética y no de una enfermedad> y es perseguido en África hasta el día de hoy según denuncia Amnistía Internacional, para concienciar sobre el sufrimiento de las personas con albinismo en África, donde no solo combaten el cáncer por la falta de pigmentación, sino también la discriminación y la persecución. Recientemente, esta asociación ha denunciado los secuestros, homicidios o mutilaciones que sufren en Malawi, donde sus cuerpos son empleados en prácticas de brujería. Resulta un buen ejemplo de cómo una enfermedad o una alteración sin graves repercusiones físicas puede llegar a ser un terrible tormento para quien la padece tan solo por nuestra educación, sociedad o cultura, cuando es incapaz de comprender realmente lo que ve, dejando que el mecanismo del miedo aumenten el daño. Síndrome de Proteus (CIE-10: Q87.3 ORPHA:744) Sin duda alguna, de entre todas las ER documentadas, existe una reconocible en todo el planeta desde hace ya más de un cuarto de siglo y que allanó el camino para ayudar a comprender este problema y difundir el drama de padecer una enfermedad extremadamente rara, gracias a la impresionante obra cinematográfica de David Lynch: El hombre Elefante (1980). 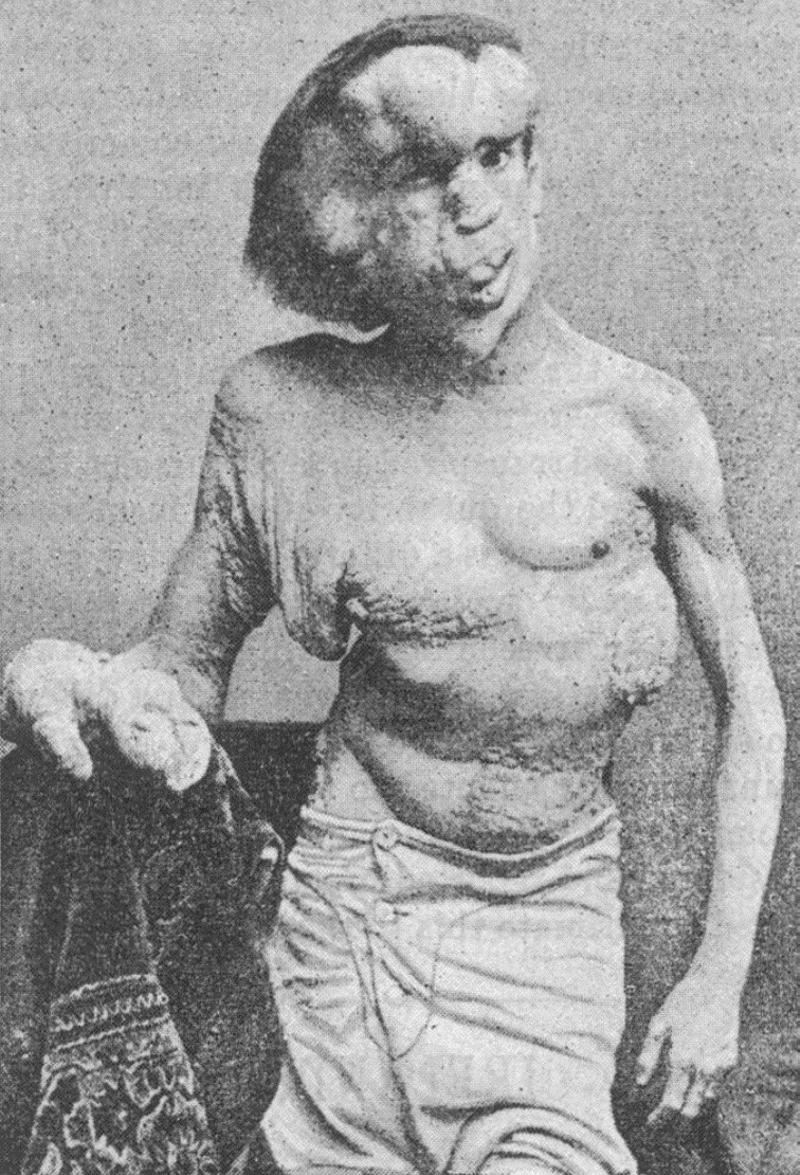 El caso de Joseph Merrick (1862 – 1890) resulta ser el más famoso de esta enfermedad realmente rara con una prevalencia de 1 caso por millón de habitantes y que desde su descubrimiento apenas se han confirmado poco más de 200 casos a finales de los años 70 del pasado siglo. Joseph Merrick fue un pobre ciudadano británico que pasó a la historia con el sobrenombre de "El Hombre Elefante", debido a que padeció uno de los casos más extremos de Síndrome de Proteus, durante la era victoriana. En un diagnóstico inicial se creyó que Merrick padecía neurofibromatosis (un trastorno genético del sistema nervioso que afecta principalmente al desarrollo y crecimiento de los tejidos de las células nerviosas), aunque con el paso de las décadas y una vez descubierto el síndrome por el Dr. Michael Cohen en el año 1979, gracias a modernas pruebas realizadas al esqueleto de Joseph Merrick se pudo comprobar que la enfermedad que realmente padeció fue dicho síndrome. Como en muchos otras ocasiones, la falta del avance científico y médico, así como que ambas enfermedades tienen síntomas parecidos, como las manchas de color café por todo el cuerpo y las grande tumoraciones, llevó a un primer diagnóstico erróneo y hubo que esperar hasta finales de los años 70 del siglo XX para conocer la verdadera naturaleza de una enfermedad que le supuso un auténtico calvario y no solo físico, si no la crueldad y burla de una sociedad poco comprensiva y solidaria con la desgracia humana. En el caso del desafortunado Merrick las consecuencias de la enfermedad provocaron que su brazo y mano derechos, así como ambas piernas, presentaran un desarrollo anormalmente grande y deforme, mientras que su brazo izquierdo tuviera un aspecto normal pero falto de desarrollo como el de un niño de entre 10 y 12 años. Desde un punto de vista médico, el Síndrome de Proteus causa un crecimiento anormal de la piel, huesos, músculos, tejido adiposo y vasos sanguíneos y linfáticos. El cuerpo puede adquirir una terrible deformidad estructural causada porque algunas partes del cuerpo se desarrollan más que otras, provocando un crecimiento exagerado en unas extremidades o falta de desarrollo en otras. Es un síndrome genético pero no hereditario, con unas consecuencias muy variables de un paciente a otro. Esta causado por una mutación genética del gen AKT1 ubicado a lo largo del cromosoma 14. A día de hoy la enfermedad se sigue investigando y se desconoce exactamente a qué cromosoma o cromosomas implica, así como el gen y el locus (posición fija en un cromosoma, que determina la posición de un gen o de un marcador genético). Se sabe que el fenotipo (la información genética que posee un organismo en particular en forma de ADN) de las personas que padecen esta enfermedad cambia con la edad, pero afecta por igual a ambos sexos, siendo autosómica recesiva (significa que un individuo debe recibir el alelo mutado de ambos padres para heredar la enfermedad). Desafortunadamente es una enfermedad progresiva sin que por el momento se disponga de una cura, siendo un buen ejemplo de las consecuencias de padecer una ER pese a ser tan conocida, pues es paradigmática en muchos aspectos, por un lado, aunque afortunadamente, puede ser diagnosticada con relativa facilidad (aunque sólo puede diagnosticarse por la observación clínica, la evolución del paciente y pruebas específicas al tener sospechas, pero con el problema agravante de que todos los signos de la enfermedad no aparecen en un mismo individuo ya que presenta una alta variabilidad de un individuo a otro). Por otro lado el tratamiento es paliativo consistiendo en la resección de los tumores deformantes y la amputación de miembros exageradamente desarrollados o malformados. Además los afectados padecen trastornos cardiovasculares que deben ser tratados farmacológicamente. Si por desgracia presentan neoplasias, (una formación anormal en alguna parte del cuerpo de un tejido nuevo de carácter tumoral, benigno o maligno) probablemente se acortará la esperanza de vida. Los niños que se ven afectados con este síndrome acostumbran a nacer sin ninguna deformidad evidente, pero la alta variabilidad de los síntomas puede llevar a que incluso algunos recién nacidos ya presenten leves malformaciones. Aunque lo común en todos los casos es que hacia año y medio de edad aproximadamente, aparezcan los primeros tumores o el crecimiento de la piel y de los huesos, así como las citadas manchas de color café con leche. Como ya hemos comentado la gravedad y la localización de estos crecimientos asimétricos varían ampliamente según los casos, pero resulta común su localización en el cráneo, columna vertebral, pelvis, uno o más miembros, dedos y en las plantas de los pies. Según el paciente en concreto, este desarrollo alterado puede detenerse al terminar la pubertad, aunque los tumores continuarán probablemente, así pues el pronóstico podrá variar ampliamente en cualquier caso, dependiendo de cómo afecte específicamente la enfermedad. Ante este panorama, actualmente el principal objetivo consiste en mejorar la calidad de vida del enfermo, lo cual debe incluir asistencia psicológica necesariamente dentro del tratamiento, ya que es una enfermedad altamente deformante y el paciente necesitará mucha ayuda para enfrentarse a ella y poder realizar una vida relativamente normal. Aunque parezca increíble, a día de hoy existen multitud de ERs con síntomas tan dramáticos o más que las citadas y que incluso no son correctamente diagnosticadas, pensemos que en este reportaje hemos citado tan solo enfermedades con un mínimo de centenares de casos conocidos, (pronto publicaremos un texto sobre las enfermedades raras documentadas con menos casos existentes, que pueden llegar incluso a un solo individuo o familia, pero que pueden ser tan mortales a corto plazo como un cáncer o una embolia cerebral ) pues existen muchas enfermedades que apenas si se conocen uno o varios casos dentro de una familia y cuyas víctimas pueden llegar a fallecer ante la incomprensión social y médica. Si a esto añadimos la precariedad económica de muchos sistemas sanitarios públicos para atender a las personas más necesitadas en muchos lugares del planeta, el problema de los afectados se multiplica con creces. Por todo ello, es importante comprender que es realmente una ER y el sufrimiento que puede acarrear tanto a pacientes como al entorno familiar ante la incomprensión y falta de solidaridad de una sociedad cada vez más volcada en la competitividad y el individualismo, incapaz de comprender que solo podremos avanzar como especie unidos y desvelando los secretos de nuestra naturaleza desde la justicia y la solidaridad. Manuel Castelló (kasmangou)
Temas relacionados: Kasmangou, Medicina , Sociedad, Economía social, Biomedicina Reconocimientos y más información sobre la obra gráfica ADVERTENCIA: En este foro, no se admitirán por ninguna razón el lenguaje soez y las descalificaciones de ningún tipo. Se valorará ante todo la buena educación y el rigor sobre el tema a tratar, así que nos enorgullece reconocer que rechazaremos cualquier comentario fuera de lugar.
2 Comentarios
Ricardo Soria
18/2/2018 21:49:15
Felicidades!!! es la primera vez que leo un artículo en Internet fuera de un espacio médico o especializado, como organizaciones en defensa de los pacientes, donde se haga una diferenciación racional entre ER y síndromes varios. Como profesional me esperaba las típicas fotos que dan repelús, textos ridículos y prescindibles... confieso que no comprendía el nombre de su página al llegar al artículo desde una búsqueda. Sinceramente sorprendido por como critican a los artículos generales magníficando síntomas estrafalarios o imágenes morbosas. Les sugiero indagen en enfermedades raras menos conocidas, el espectro es muy amplio y desconocido, al igual que sus síntomas y el drama que supone para los afectados, y como acertadamente mencionan, no me refiero a los casos de hipocondría que tanto destacan las noticias y daño hacen a la credibilidad de los auténticos pacientes. Hacía tiempo que no encontraba en la red un sitio tan heterodoxo en sus contenidos, por decirlo de alguna manera. Hagan el favor de no desaparecer o no empezar a vender productos milagro si se mantienen, este esquema es muy distinto a todo lo visto ¿Donde está el truco?
Quelecortenlacabeza
18/2/2018 22:51:56
He leído sobre algunos de estos síndromes más de una vez, pese a no ser enfermedades, incluso en la televisión e informativos de audiencia como si fueran las más raras y me extrañaba muchísimo... era imposible, aunque no veía la diferencia, ahora lo entiendo mejor. Como pone Ricardo Soria, podríais explicar una lista con las realmente más únicas, pude acceder a una lista a través de un enlace que pusisteis de Orphanet pero no comprendía nada y la Wikipedia no te aclara de que van, sus nombres pico tienen que ver con esos tan espectaculares que ponéis de los síndromes que ponen por la tele. Tengo curiosidad. Gracias Deja una respuesta. |


Investigación Médica y Salud
Cine con conciencia
Educación y Formación



Cultura y Ocio

¿Tienes una cita?
Lucha por unas redes sociales y un Internet seguro para la infancia
Colabora para impedir la violencia de género

Si crees que puedes ser víctima, no dudes en llamar, tu vida es lo primero! |
