

 La tendencia acentuada de los medios por acaparar la audiencia, ha ido reemplazando la información sobre las ERs y su investigación por una sucesión de espectáculos escabrosos o testimonios sensacionalistas más propios un programa del corazón. Coincidiendo con el día internacional de las enfermedades raras vamos a mostraros y analizar las enfermedades más únicas del mundo, que como ya os explicamos en un anterior reportaje, poco tienen que ver con las mostradas habitualmente en los medios de comunicación, donde se tiende a buscar los casos con los síntomas más espectaculares y las situaciones más morbosas. Desafortunadamente la tendencia cada vez más acentuada de los medios de comunicación por acaparar la audiencia a cualquier precio, ha ido paulatinamente reemplazando la información sobre las enfermedades raras o los casos más únicos y su investigación (o ausencia de ella) por una sucesión de espectáculos escabrosos o bien directamente de relatos y testimonios sensacionalistas que parecen directamente sacados de un programa del corazón para obtener la lágrima fácil y el asombro que mejore los ingresos publicitarios de un público ávido de presenciar el sufrimiento ajeno. En un día como hoy esa clase de programas y artículos bochornosos se multiplicará mostrando toda clase de síndromes extraños, que muchas veces no son ni siquiera enfermedades raras o quizás, relatos en primera persona que garanticen una buena ración de sufrimiento ajeno, sin importar el rigor científico o la más mínima deontología periodística, tan solo se trata de aprovechar el momento, olvidando la reflexión profunda objeto de este día, y eso considerando que decidan otorgarle la correspondiente relevancia mientras no dañe publicidad, audiencia en ingresos. Pero los pacientes de ERs (Enfermedades Raras) existen los 365 día del año, no solo hoy, así que esta clase de pseudo - periodismo interesado muchas veces hace un daño irreparable que nos aleja de poder comprender o ayudar a este gran colectivo del cual muchos medios solo se acuerdan según dicta el calendario o el drama de turno que aumente sus ingresos etiquetándolo de la manera adecuada.  Las ERs, ya lo hemos dicho muchas veces, no resultan interesantes para gran parte de la industria médica y exigen por parte de los gobernantes un esfuerzo de solidaridad y medios que rara vez están dispuestos a afrontar en ausencia de un rédito electoral cortoplacista. Así que dado el día que es, por supuesto también hoy hablaremos de ERs (pero no solo hoy, procuramos hacerlo habitualmente y con este reportaje, tratar en sucesivos trabajos la totalidad de las ERs actualmente reconocidas ) así que vamos a intentar que se conozcan las realmente más raras y extrañas desde un punto de vista médico y científico, además de intentar explicar sus síntomas, características, consecuencias o esperanzas en el campo de la investigación como el problema real, de compleja solución como son, si no atendemos a la solidaridad y la comprensión que requiere un drama del que podemos ser partícipes en cualquier momento. Ya mostramos en anteriores reportajes como rara vez coincide lo que los medios muestran como una enfermedad realmente muy rara y la realidad, así mucho menos, el problema de los procesos diagnósticos, la impotencia ante la falta de fármacos y tratamientos o el sufrimiento añadido que supone para una familia frente a una enfermedad convencional. Hoy nos vamos a centrar en los casos más únicos para la ciencia, lo cual quiere decir que tan sólo se ha confirmado un caso o a lo sumo, se han dado casos en una sola familia. Invitamos a nuestros lectores a que comparen la información que aquí se presenta hoy con lo que habitualmente se muestra y como se muestra, comprobará que quizás más de un periodista a perdido el norte (o quizás más bien a encontrado su vocación en el campo de la economía). Como ya explicamos anteriormente, existen varias instituciones y organismos que realizan informes rigurosos sobre la prevalencia de las enfermedades más raras, uno de los estudios más relevantes lo realiza Orphanet, organización establecida en Francia en 1997 y que con la implantación de internet, buscó reunir los escasos conocimientos sobre ERs con el fin de mejorar el diagnóstico, la atención y el tratamiento de los pacientes con dichas enfermedades. Con el éxito obtenido esta iniciativa pronto se convirtió en un empeño europeo en el año 2000, apoyado por la Comisión Europea, Orphanet ha crecido gradualmente hasta constituir un Consorcio de 40 países, dentro de Europa y en todo el mundo. Durante los últimos 20 años, Orphanet se ha transformado en una de las fuentes de información de referencia, facilitando el acceso a todas las audiencias desde profesionales sanitaros e investigadores hasta familiares y afectados con datos de calidad entre la plétora de información disponible en línea, ofreciendo los medios para identificar a los pacientes con enfermedades raras y contribuir a generar conocimiento mediante la producción de datos científicos masivos, computables y reutilizables. Uno de los informes más curiosos e interesantes que realizan es el referido a la prevalencia de las diferentes ER, del cual vamos a partir basar esta serie de trabajos. Aunque Orphanet basa su trabajo en la vigilancia sistemática de la literatura médica con el fin de estimar la prevalencia e incidencia de las enfermedades raras. El citado estudio, con una prioridad bianual tiene como objetivo recopilar nuevos datos respecto a la prevalencia puntual, la prevalencia al nacimiento y la incidencia, actualizando los datos ya publicados previamente según los nuevos estudios científicos, informes sanitarios y otros datos relevantes disponibles. Actualmente hay 5653 enfermedades raras con información sobre prevalencia e incidencia en la base de datos de Orphanet, pero este estudio parte de presentar las enfermedades o grupos de enfermedades de diversas maneras: por un orden decreciente de prevalencia, de incidencia o por el número decreciente de casos o familias publicados. Se parte de una prevalencia de menos de 1000 casos a nivel mundial y en otra de 100, es decir entre 1 y 100 afectados por una de estas enfermedades entre más de 7.500 millones de seres humanos. ¿Por qué son necesarios estos diferentes métodos? Para poder clasificar las enfermedades (aunque el método sería extrapolable a otras organizaciones y agencias interesadas en la materia) esta organización europea utiliza toda una serie de diferentes fuentes de información desde las distintas base de datos con registros especializados como RARECARE o EUROCAT a la información suministrada por los distintos institutos y agencias sanitarias nacionales e internacionales que se interesan por las ERs por diversas razones: Institut National de Veille Sanitaire, American Center of Disease Control and Prevention, American National Cancer Institute, Agencia Europea de Medicamentos, Organización Mundial de la Salud, etc. Pero además y aquí es donde está la verdadera “fuerza” de este método, se tratan informáticamente y se cruzan los de datos epidemiológicos de Medline, probablemente la base de datos de bibliografía médica más amplia que existe en el mundo, realizada por la Biblioteca Nacional de Medicina de los Estados Unidos y que en realidad es la versión evolucionada e informatizada a de tres base de datos: Index Medicus, Index to Dental Literature e International Nursing Index.  Diferentes servicios ofrecidos por Orpha.net A su vez toda esta información se cruza con toda clase de textos médicos e informes de expertos que salen a la luz por diversos medios y que son analizados por los expertos que colaboran con Orphanet alrededor del planeta. Aún y así, los datos publicados en estas listas son estimaciones mundiales o estimaciones europeas si no hay disponible una estimación a nivel mundial, pues resulta realmente difícil realizar una estimación real y clara de la incidencia de muchas enfermedades por la falta de colaboración o recursos de muchos países y gobiernos. Aunque parezca increíble, a nivel mundial todavía existen materias básicas como son la sanidad donde las naciones no se ponen de acuerdo y donde la política o intereses egoístas prevalecen sobre el interés del bienestar y la salud general. Así pues, los datos publicados son información recopilada en bruto, extrapolaciones de datos en bruto a nivel mundial o europeo cuando no se sospecha un efecto fundador como causa de una enfermedad. Cuando se dispone de una serie de datos nacionales, se calcula la media para estimar la prevalencia o incidencia mundial o europea. En el caso de disponer de una variedad de fuentes de datos que entran en contradicción, siempre prevalece la fuente de información más reciente, siempre que sus criterios de calidad y rigor sean validos.  Los medicamentos llamados huérfanos son todos los productos con propósitos medicinales que sirven para diagnosticar, prevenir o tratar enfermedades y desordenes que puedan amenazar la vida y salud pero que son raros. Se le así porque la industria farmacéutica tiene poco interés en ellos, bajo las condiciones normales del mercado, pues para desarrollar y poner en el mercado estos productos, dirigidos solamente a una pequeña cantidad de pacientes, las probabilidades de recuperar la inversión y conseguir beneficios son muy bajas. Promover por tanto, la investigación para el conocimiento de las causas de las enfermedades raras, su tratamiento, el desarrollo de fármacos, así como la investigación en ciencias sociales para mejorar el conocimiento de la situación social, sanitaria, laboral y educativa del colectivo y detectar las necesidades no cubiertas de las personas con ER es una de las misiones fundamentales promovidas por las grandes organizaciones de afectados. ¿Cómo se estima la prevalencia? En enfermedades congénitas, la prevalencia se estima, calculando la prevalencia al nacimiento por la expectativa de vida del paciente dividido por la expectativa de vida de la población general Pero cuando sólo están documentados los datos de incidencia, la prevalencia se estima si es posible, multiplicando la incidencia por la duración media de la enfermedad. Ahora bien, en el caso de enfermedades muy raras y poco frecuentes, sin datos de prevalencia ni de incidencia, sencillamente se ofrece el número de casos o de familias documentadas en la literatura. Los datos de prevalencia e incidencia en estos informes, como bien los propios creadores insisten, son tan solo estimaciones y no pueden ser considerados absolutamente correctos. Todavía estamos muy lejos de disponer a nivel mundial de un sistema colaborativo que permita analizar con absoluta precisión los datos epidemiológicos, aunque el esfuerzo de esta organización por conseguir resultados válidos es destacado a nivel mundial por la comunidad científica.  Con este reportaje, no solo pretendemos hacer un recordatorio del problema y el reto que representan las enfermedades raras, si no que deseamos alejarnos de las habituales reseñas simplistas o espectaculares que nos apartan de lo que realmente son: Una compleja realidad médica y social, terrible y apasionante a la vez, donde la ciencia y la solidaridad van unidas de la mano y deben ser tratadas con el rigor y respeto que los afectados y nuestro lectores ávidos de conocimientos se merecen. Una vez aclarado esto, en nuestro reportaje nos vamos a centrar en los casos únicos del estudio de Orphanet, donde ni siquiera podemos hablar realmente de una incidencia o prevalencia, es decir, sencillamente personas afectadas por una enfermedad rara, con síntomas y un diagnóstico claro, pero que tan solo se conoce un caso o se han dado casos únicamente en miembros de una familia alrededor de todo el planeta, intentando usar un lenguaje claro y sencillo hasta lo posible, que nos ayude a comprender la complejidad y variedad de enfermedades extrañas a las que nos podemos enfrentar. Ahora bien, muchas veces las ERs, son complejas familias de enfermedades, así que comentaremos todas las vinculadas a una familia concreta. Hablamos de poco más de 50 enfermedades con un caso único en el mundo, si se dan 2 casos, casi se alcanzan ya sin embargo los 500 pacientes y con 4 casos ya superamos ampliamente el millar de pacientes aquejados de una enfermedad prácticamente única, dejando claro el tremendo abanico de combinaciones posibles a las que se tiene que hacer frente. Condrodisplasia letal tipo Seller (Orpha: 1421) Condrodisplasia letal tipo Moerman (Orpha: 1420) Osteocondrodisplasia letal compleja (Orpha: 457378) Condrodisplasia recesiva letal (Orpha: 1423) Condrodisplasia punctata rizomélica (Orpha: 1420) Comenzaremos con un ejemplo claro de enfermedades mortal, sin apenas información ni opciones, son enfermedades, que como en el caso de la Condrodisplasia letal tipo Seller, conduce a una muerte temprana en menos de un año después de la aparición de los primeros síntomas o su diagnóstico. Habitualmente el término condrodisplasia hace referencia a toda una serie de de afecciones que son causadas por ciertos cambios en los genes y a menudo vinculadas al enanismo. Habitualmente impide el crecimiento de muchas partes del cuerpo, especialmente los huesos, pero las personas con algunos tipos de condrodisplasia pueden llevar una vida normal con pocas limitaciones, sin embargo otros tipos menos comunes pueden llegar a causar discapacidades físicas y mentales. En los casos extremos del espectro se encuentran las formas severas y dolorosas de condrodisplasia, como la más conocida condrodisplasia punctata rizomélica (RCDP1) enfermedad que impide el crecimiento en los bebés, causando problemas óseos, discapacidades mentales y cataratas. En estos casos la mayoría de las personas que la padecen no viven más allá de la infancia. Pero en el caso de las variantes Seller, Moerman o letal compleja su rápido desarrollo y capacidad para detener la función de los órganos les ha llevado a ser bautizadas de manera realmente excepcional en su propio nombre genérico con el adjetivo terrorífico de “letal”. 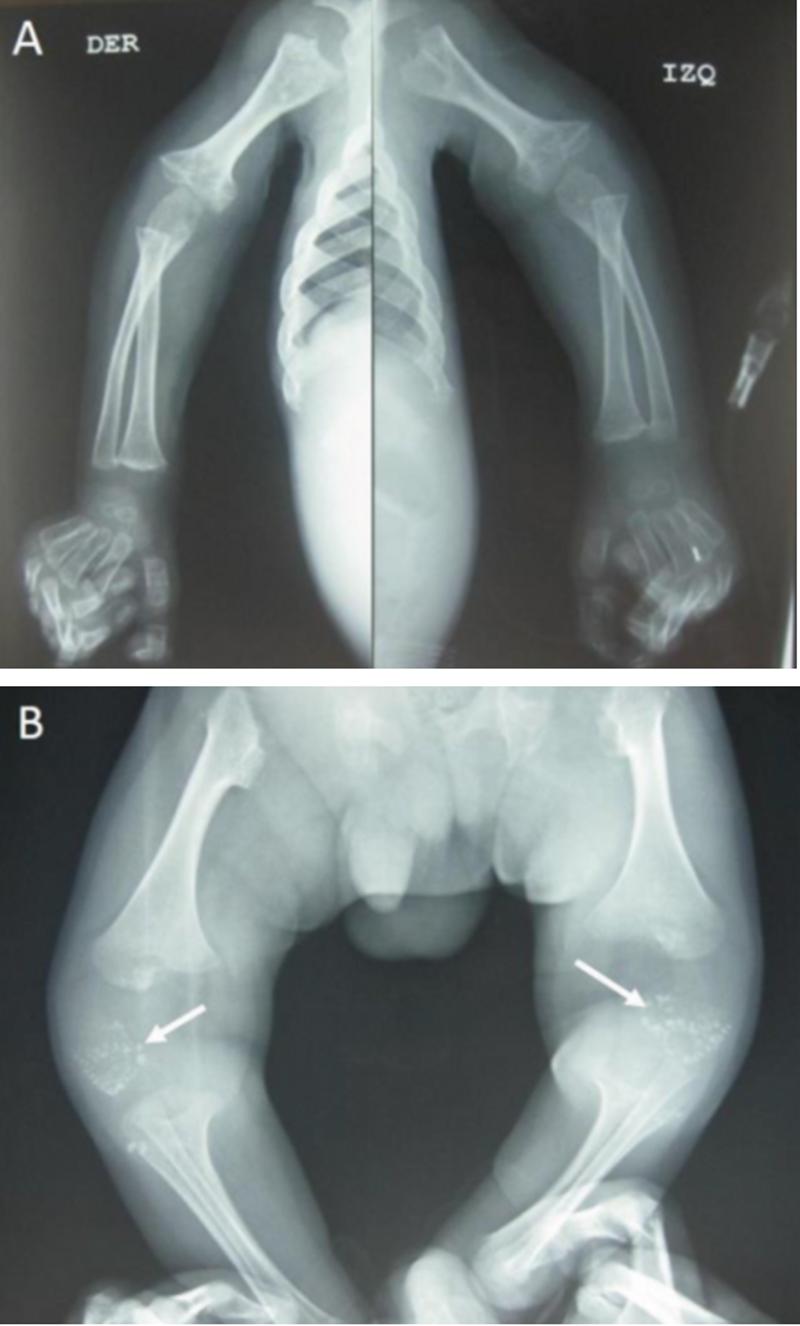 Caso clínico, de enfermedad peroxisomal y condrodisplasia rizomelica punctata tipo 1, Proyección anteroposterior comparativa de miembros. A. Ambos húmeros muestran significativo acortamiento en relación a huesos del antebrazo y ensanchamiento metafisiario. B. Calcificaciones puntiformes en rótula y epífisis distal de fémures, ensanchamiento metafisiario con acortamiento diafisiario femoral (rizomélico). Cesar Leonardo González-Ortiza, Sandra Bibiana Jaimes Leguizamónb y Gustavo Adolfo Contreras-Garcíac Las condrodisplasias son genéticas pero afortunadamente, como ya hemos advertido, solo existen muy pocos casos de las variantes más letales, por suerte los investigadores están trabajando en posibles tratamientos de al menos algunas variantes en la actualidad como por ejemplo de la condrodisplasia punctata rizomélica. Las condrodisplasias en general, por su naturaleza genética se encuentran ampliamente ligadas con las enfermedades raras y acostumbran a ser diagnósticos muy duros y abrumadores para los pacientes y familia por las condiciones de estas dolencias que conllevan duras consecuencias físicas y mentales, que pueden conducir además a la muerte en un periodo corto de tiempo. Junto a las ya mencionadas en la lista aparecen algunas más de la familia dentro de las ERs: Condrodisplasia punctata dominante ligada al X (Orpha 35173) Osteocondrodisplasia hipertricótica tipo Cantu (Orpha: 1517) Espondiloencondrodisplasia (Orpha: 1855) Condrodisplasia metafisaria tipo Spahr (Orpha: 2501) Condrodisplasia metafisaria tipo Jansen (Orpha: 33067) Condrodisplasia tipo Blomstrand (Orpha:50945) Condrodisplasia dominante ligada al X tipo Chassaing-Lacombe (Orpha: 163966) Leucoencefalopatía - condrodisplasia metafisaria (Orpha: 83629) Condrodisplasia con dislocaciones articulares tipo gPAPP (Orpha: 280586) Condrodisplasia punctata rizomélica tipo 5 (Orpha: 468717) Condrodisplasia punctata tipo Toriello (Orpha: 79347) Condrodisplasia - trastorno del desarrollo sexual (Orpha: 1422) Condrodisplasia metafisaria tipo Kaitila (Orpha: 166038) Condrodisplasia metafisaria - retinosis pigmentaria (Orpha: 166035) Inmunodeficiencia primaria con infección viral post- sarampión- paperas- rubéola (Orpha: 431166) Inmunodeficiencia por deficiencia de ficolina3 (Orpha: 331190) Inmunodeficiencia por deficiencia de MASP-2 (Orpha: 331187) Inmunodeficiencia combinada por deficiencia de OX40 (Orpha: 431149) Otra condición muy habitual en las ERs resulta ser las inmunodeficiencias, un estado patológico donde el sistema inmunitario no cumple con el papel de defensa del organismo, dejándolo vulnerable a las infecciones, así que causan a quienes las padecen una gran susceptibilidad a padecer infecciones o una mayor posibilidad de padecer como consecuencia cánceres de diversos tipos. Las inmunodeficiencias primarias, como es el caso de la Inmunodeficiencia primaria con infección viral post- sarampión- paperas- rubéola son otro antiguo conocido dentro de las enfermedades raras y existen numerosos proyectos de investigación al respecto así como asociaciones, que se engloban dentro de este marco que incluye también enfermedades autoinflamatorias y autoinmunes. Todas las inmunodeficiencias primarias (IP) son un grupo raro de enfermedades del sistema inmunitario, habitualmente hereditarias, congénitas y de origen genético, por ejemplo esta concretamente tiene ya identificados dos genes asociados: IFNAR2 (interferon alpha and beta receptor subunit 2) como factor importante de susceptibilidad en la Inmunodeficiencia primaria y el STAT2 (signal transducer and activator of transcription) responsable de las mutaciones germinales causantes de enfermedad. Las manifestaciones clínicas se inician casi siempre en algún momento de la primera infancia, con pacientes que tienen a causa de su vulnerabilidad constantes infecciones, causadas por patógenos normales, infecciones recurrentes o crónicas difíciles de erradicar e infecciones por gérmenes oportunistas o no habituales, si a esto sumamos que se produzcan tras padecer sarampión, paperas, rubéola no enfrentamos a una patología infantil muy seria y peligrosa. Todas las IPs son enfermedades muy raras cuya incidencia oscila entre 1/10.000-1/100.000 de los nacidos vivos, exceptuando la deficiencia aislada de la inmunoglubina A (IgA), que se sitúa entre 1/200-1/1.000. Aunque estos trastornos requieren una atención muy especializada, muchas veces son los pediatras o médicos de familia quienes primero ven a estos niños, con frecuencia en repetidas ocasiones, antes de ser diagnosticados. Un alto índice de sospecha puede salvar la vida del niño, pues conduce al diagnóstico y tratamiento (por ejemplo, con trasplante de progenitores hematopoyéticos) de la enfermedad. Además, permite establecer medidas profilácticas frente a infecciones oportunistas y, lo que no es menos importante, evitar actuaciones como la administración de vacunas de microorganismos vivos o sangre no irradiada, que pueden tener consecuencias mortales para el paciente. En el caso de la inmunodeficiencia debida a la deficiencia de ficolina3 es otra inmunodeficiencia rara, genética que se inicia en la infancia, debida a una anomalía en una proteína caracterizada por niveles bajos o indetectables de ficolina3 sérica. Esto hace susceptible al paciente a muchas infecciones y probablemente otros problemas de autoinmunidad. Los síntomas son variables y graves: desde enterocolitis necrosante perinatal e infecciones cutáneas recurrentes por Staphylococcus aureus hasta infecciones pulmonares recurrentes de aparición en la infancia que conducen a abscesos cerebrales y fibrosis pulmonar a nefropatía membranosa.  En general como hemos visto todas estas inmunodeficiencias a las que hacemos referencia y que generalmente desde la primera infancia producen “puertas” a toda clase de infecciones y cáncer, pero si además hablamos de una inmunodeficiencia extraña o tardíamente diagnosticada, puede llevar con facilidad a consecuencias fatales para el paciente. Son enfermedades graves que pueden presentar con facilidad múltiples complicaciones al dejarnos expuestos y sin defensas Dentro de esta familia de ER se encuentran un amplio listado dentro de las más excepcionales con tan solo unos pocos casos por ER que os ponemos a continuación:
Síndrome orofaciodigital tipo 12 (Orpha: 141327) Síndrome orofaciodigital tipo 13 (141330) Dentro de las ERs más raras que afectan a la constitución física visible nos encontramos con el síndrome oralfaciodigital, que es en realidad un grupo de afecciones relacionadas entre sí y que afectan el desarrollo de la cavidad oral, es decir la boca y los dientes, así como las características faciales y los dedos tanto de las manos como de los pies. Los investigadores han identificado al menos 14 formas potenciales de este síndrome que se clasifican por sus patrones de signos y síntomas. Sin embargo, las características de los distintos tipos se superponen significativamente, y algunos tipos no están bien definidos. El sistema de clasificación para el síndrome oral-facial-digital continúa evolucionando a medida que los investigadores encuentran más personas afectadas y aprenden más sobre este trastorno. Los signos y síntomas del síndrome varían ampliamente según el tipo, pero todos conllevan graves problemas con el desarrollo de la cavidad oral, los rasgos faciales o los dedos, pero lo peor de todo es que la mayoría de las formas de este síndrome también están asociadas con anormalias cerebrales y algún grado de discapacidad intelectual. Las anomalías de la cavidad oral que ocurren en la mayor parte de los casos de este síndrome incluyen una división (hendidura) en la lengua , con una forma lobulada inusual y el crecimiento de tumores o nódulos no cancerosos en la lengua. Las personas afectadas también pueden llegar a tener dientes extra, faltantes o defectuosos, así como una abertura en el techo de la boca, es decir un paladar hendido, así como bandas de tejido adicional (llamado frenula hiperplásica) que unen anormalmente el labio a las encías. Las anomalías de los dígitales incluyen la fusión de ciertos dedos , dedos más cortos de lo normal (braquidactilia) o inusualmente curvos (clinodactilia) También puede ocurrir la presencia de dedos adicionales (polidactilia) . 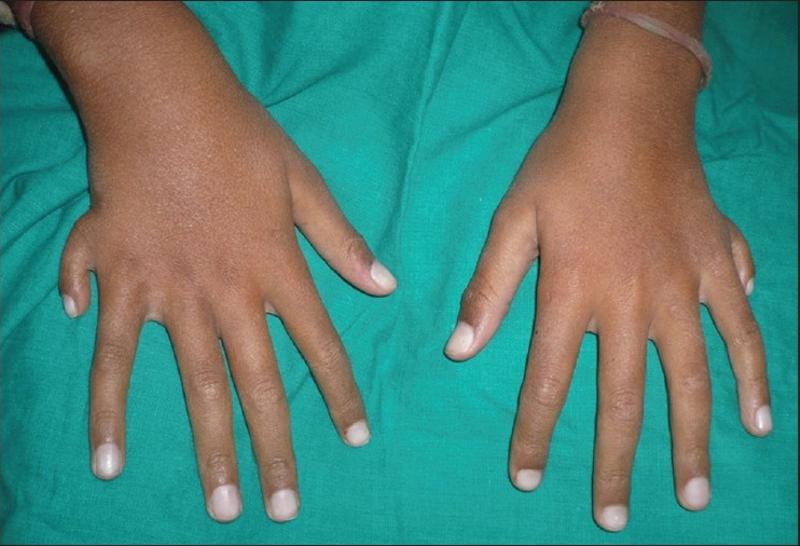 Caso de polidactilia en ambas manos asociado al Síndrome orofaciodigital. Indian Journal of Dental Research Las diferentes características ayudan a distinguir las distintas formas del trastorno. Por ejemplo, la forma más común de síndrome tipo I, está asociada con la enfermedad renal poliquística, que se caracteriza por el crecimiento de sacos llenos de líquido (quistes) que interfieren con la capacidad de los riñones para filtrar productos de desecho de la sangre. Otras formas se caracterizan por problemas neurológicos, cambios particulares en la estructura del cerebro, anomalías óseas, pérdida de visión y defectos cardíacos. Otras variantes presentes en el listado con algunos pocos casos más serian:
Deficiencia neonatal transitoria múltiple de acil-CoA deshidrogenasa (Orpha: 329942) Por suerte no todas las ER tienen un diagnóstico fatal y a veces se obtiene una solución, incluso en este caso único en el mundo donde se pudo resolver este extraño déficit. La deficiencia neonatal transitoria múltiple de acil CoA-deshidrogenasa es una enfermedad rara en la que un déficit de riboflavina materna provoca en el lactante manifestaciones similares a las observadas en la deficiencia múltiple de acil CoA-deshidrogenasa (MADD) (otra ER donde se produce una alteración de la oxidación de los ácidos grasos y los aminoácidos y un trastorno clínicamente heterogéneo que va desde la presentación neonatal grave con acidosis metabólica, miocardiopatía y hepatopatía, hasta una enfermedad leve de la infancia o la edad adulta con descompensación metabólica episódica, debilidad muscular e insuficiencia respiratoria.) En este caso, el único caso descrito en el mundo, la madre del niño afectado presentaba haploinsuficiencia del transportador humano de la riboflavina (hRFT1) y esta deficiencia natal se presentó como dificultad para la succión, acidosis metabólica e hipoglucemia, pero que se resolvió completamente gracias a la administración oral de riboflavina. Paraplejía espástica autosómica recesiva tipo 60 (Orpha: 401800) Paraplejía espástica autosómica recesiva tipo 68 (Orpha: 401825) Paraplejía espástica autosómica recesiva tipo 71 (Orpha: 401840) Las paraplejias espásticas hereditarias (HSP) comprenden un grupo genética y clínicamente heterogéneo de trastornos neurodegenerativos caracterizados por una espasticidad progresiva (trastorno motor del sistema nervioso en el que algunos músculos se mantienen permanentemente contraídos, contracción que provoca la rigidez y acortamiento de los músculos e interfiere sus distintos movimientos y funciones como la manipulación o el equilibrio) así como hiperreflexia (aumento de los reflejos osteotendinosos) de las extremidades inferiores. Se cree que las HSP en general, afectan a 1 de cada 20.000 individuos de la población general europea, es decir, una incidencia relativamente alta.  La fisioterapia y la rehabilitación física es extremadamente importante para mejorar el rango de movimiento de las extremidades afectada, así como el fortalecimiento de los músculos.. En la imagen podemos ver un paciente que sufre de paraplejia espástica hereditaria la cual le provoca un alto grado de espasticidad y debilidad en los músculos de las piernas por lo que resulta difícil incluso ponerse de pie. Texum Clínicamente, las HSP pueden dividirse en dos grupos principales: formas puras y complejas. Las puras se caracterizan por espasticidad y debilidad progresiva de las extremidades inferiores, asociadas a menudo con trastornos urinarios hipertónicos, una leve reducción de la sensibilidad a las vibraciones y, ocasionalmente, de la percepción de la posición de las articulaciones. Las complejas por su parte, más problemáticas, se caracterizan por la presencia de otros síntomas neurológicos y no neurológicos. La HSP puede heredarse de forma autosómica dominante, autosómica recesiva, como es el caso de las variantes más extrañas o recesiva ligada al cromosoma X. A día de hoy las HSP son todavía un gigantesco puzle por resolver presentándose múltiples formas recesivas y dominantes.. Hasta la fecha, se han localizado 31 loci diferentes responsables de las HSP puras y complejas. A pesar del número creciente de loci identificados, solo se han identificado 11 genes autosómicos y 2 ligados al X, por lo que la base genética concreta para la mayoría de las HSP está aún por determinar. El diagnóstico para poder identificar un tipo concreto se basa en el examen clínicos y toda una serie de exploraciones adicionales como electroencefalogramas, técnicas de imagen, determinación del nivel de ácidos grasos de cadena larga, electromiograma y serología para el virus linfotrópico humano de los linfocitos T (HTLVI). Es un proceso largo y costoso donde para completar el diagnóstico diferencial de este síndrome se requieren aún más pruebas de concreción (esclerosis múltiple, deficiencia de vitamina B12, distonía con respuesta a dopaminérgicos, esclerosis lateralamiotrófica o primaria, parálisis espástica ascendente hereditaria y paraplejía espástica provocada por infección de HTLVI). Además de las variantes nombradas, en la lista figuran casi medio centenar de paraplejias espásticas con un número de casos que suele oscilar entre 2 y el centenar de forma autosómica dominante, autosómica recesiva o recesiva ligada al cromosoma X pero además podemos encontrar también las siguientes variantes: Paraplejía espástica - glaucoma - discapacidad intelectual (Orpha: 2818) Paraplejía espástica - enfermedad ósea de Paget (Orpha: 329475) Paraplejía espástica - neuropatía – poiquilodermia (Orpha: 444099) Paraplejía espástica hereditaria (Orpha: 320365) Con esto terminamos este primer reportaje sobre las ERs más singulares desde un plano científico, que esperamos ayude a comprender mejor la compleja batalla que es para la ciencia las ERs y el duro drama para los afectados, que además podríamos ser cualquiera de nosotros en algún momento de nuestra vida, donde la concienciación y la solidaridad son vitales para poder ayudar a resolver este gran reto del ser humano.  Manuel Castelló (kasmangou)
Temas relacionados: Kasmangou, Medicina , Sociedad, Economía social, Biomedicina Reconocimientos y más información sobre la obra gráfica ADVERTENCIA: En este foro, no se admitirán por ninguna razón el lenguaje soez y las descalificaciones de ningún tipo. Se valorará ante todo la buena educación y el rigor sobre el tema a tratar, así que nos enorgullece reconocer que rechazaremos cualquier comentario fuera de lugar.
3 Comentarios
28/2/2018 20:51:21
Me gustaría que pudieran ayudarme a investigar sobre el sudeck y que se dé visibilidad,para que les reconozcan sus derechos , a quienes lo padecen cada día
Manuel Castelló (admin)
28/2/2018 21:05:49
Ok Noelia, tomamos nota sobre, síndrome del dolor regional complejo, haremos lo que podamos, en serio. Ánimo. Tendrás noticias nuestras Deja una respuesta. |


Investigación Médica y Salud
Cine con conciencia
Educación y Formación



Cultura y Ocio

¿Tienes una cita?
Lucha por unas redes sociales y un Internet seguro para la infancia
Colabora para impedir la violencia de género

Si crees que puedes ser víctima, no dudes en llamar, tu vida es lo primero! |
